
Translate this page into:
Osteogenesis imperfecta: Strengthening knowledge of bones and genes

*Corresponding author: Shubha R Phadke, Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow, Uttar Pradesh, India. shubharaophadke@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Phadke SR. Osteogenesis imperfecta: Strengthening knowledge of bones and genes. J Pediatr Endocrinol Diabetes 2023;3:50-7.
Abstract
Twenty-two disorders associated with recurrent fractures and decreased bone density caused by 20 genes are grouped under osteogenesis imperfecta. Identification of causative genes has strengthened the understanding of bone formation and mineralization. The mode of inheritance can be autosomal dominant, recessive, or X-linked. Genetic diagnosis is essential for genetic counseling. The presentation can be from fetal life to adult life, and suspicion in appropriate clinical settings is essential for ordering the investigations. This challenging task of identifying the causative genetic variant of such an etiologically heterogeneous group of disorders is simplified by the high throughput sequencing technique of next-generation sequencing. In this era, each case needs to be diagnosed by DNA-based methods. Genetic counseling and prenatal diagnosis are important parts of management. The evolving novel therapies are likely to be specific to the disease mechanisms and will supplement the current treatment with bisphosphonates and better outcomes are expected in the future.
Keywords
Osteogenesis imperfecta
COL1A1
COL1A2
Genetic diagnosis
Genetic counseling

INTRODUCTION
Osteogenesis imperfecta (OI) is a group of single-gene disorders associated with decreased bone density and recurrent fractures with or without other features such as blue sclera, dentinogenesis imperfecta, and deafness. There is great phenotypic and genetic heterogeneity. Most of the cases are caused by heterozygous mutations in COL1A1 or COL1A2 genes [Figure 1a and b]. Despite identifying these causative genes in the 1980s; the etiology of the remaining 10% of cases continued to remain unknown for a long time. Recurrences in the families with severe types of OI such as type II or III were attributed to germline mosaicism till CRTAP, the first gene causing autosomal recessive OI was identified in 2006.[1] Since then, especially in the past 10 years, 16 more genes of recessive OI have been identified. Autosomal recessive OI appears to be more common in Indian patients as compared to the Western data accounting for about 30% of OI. The powerful technique of next-generation sequencing (NGS) has contributed greatly to the identification of novel genes for OI and the description of the vast spectrum of causative variants in these genes. The information on genes for OI has provided an opportunity to peep into the process of bone mineralization and osteoporosis. The improved understanding will add to the unmet needs of therapeutic strategies for better outcomes. There is a large amount of data about the genotypic and phenotypic spectrum of OI and the use of bisphosphonates (BPs) in the management of OI. Identification of causative genetic variants is important as the risk of recurrence in the siblings may vary from 1% to 50%. Genetic diagnosis helps in genetic counseling and prenatal diagnosis which helps families, especially with severe phenotypes.
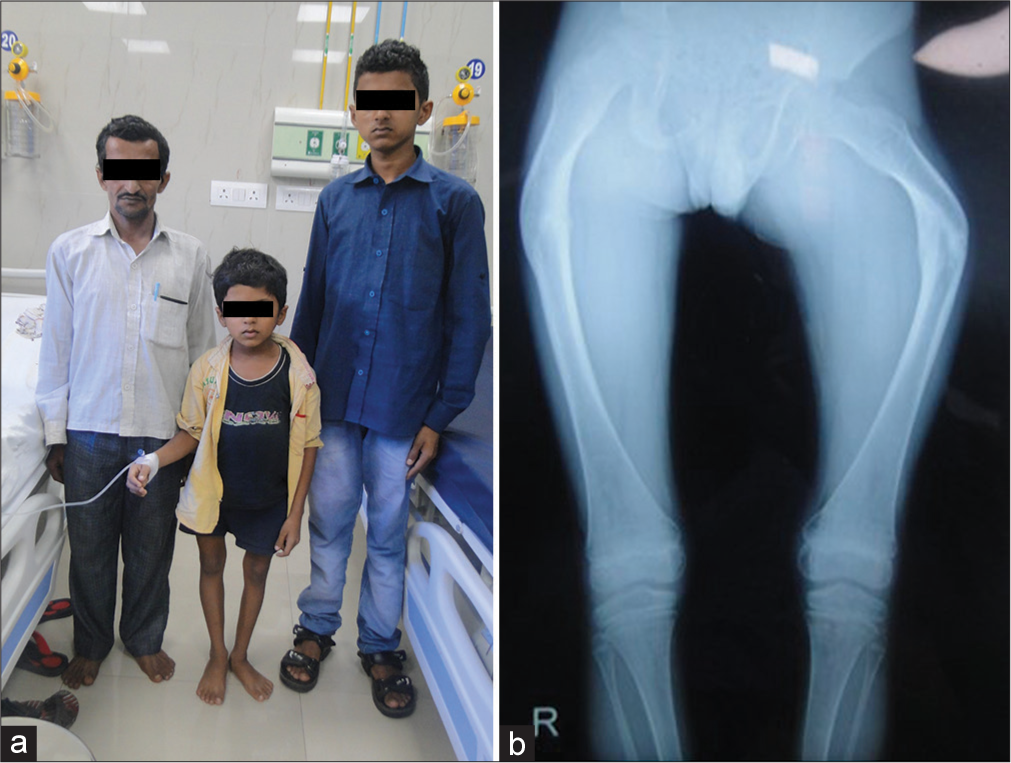
- A family of osteogenesis imperfecta type IV due to pathogenic variant in COL1A2 gene (a) Affected father and his two sons (b) Bent femora.
With its genetic and phenotypic heterogeneity, OI is a prototype of learning monogenic diseases, the use of NGS-based diagnosis, genetic counseling, and the development of medicines based on the understanding of pathogenesis. It highlights how strong molecular knowledge obtained by research on weak bones impacts clinical medicine in a short time.
ETIOLOGY, PATHOGENESIS, AND PHENOTYPES
Clinical features
OI or “brittle bone disease” is a genetically and clinically heterogeneous group of heritable single-gene disorders of connective tissue characterized by common features of recurrent fractures, usually short stature, brittle opalescent teeth, blue sclera, and adult-onset hearing loss [Figure 2a and b]. It is a common type of skeletal dysplasia accounting for 11% of patients with genetic bone disorders from India.[2] OI was also found to be the second-most common skeletal dysplasia next to the achondroplasia and also accounting for 6% of children presenting with short stature to a medical genetics department.[3] Out of the eight children in this study six had height <−4 SD indicating short stature to be an important part of the phenotype. The severity varies greatly and many cases are stillborn or die during neonatal period or infancy. The phenotype-based classification by Sillence has been in use for more than 4 decades and continues to be used to classify the phenotypes.[4] Sillence classification highlights the stark variability of the phenotypes.
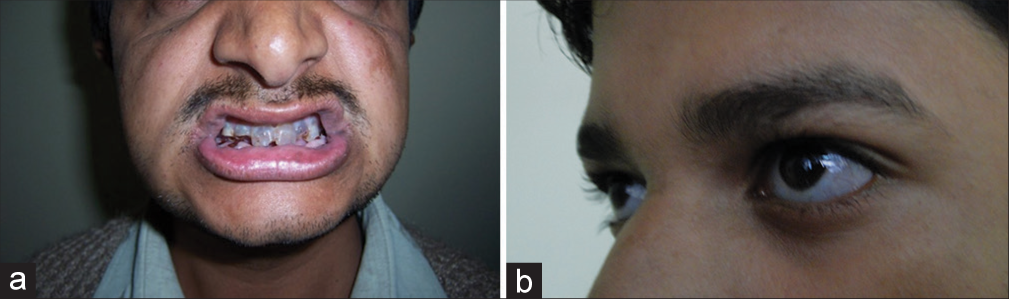
- (a) Dentinogenous imperfecta (b) Bluish sclera.
Type II is a lethal variety and can be easily detected by prenatal ultrasonography (USG) around 20 weeks of gestation. Although the presence of crumpled, bent short bones, narrow thorax, and improved visibility of intracranial structures due to decreased bone density gives clues to the diagnosis of OI, the confirmation of diagnosis needs to be done by postnatal clinical and radiological evaluation [Figure 3a and b].
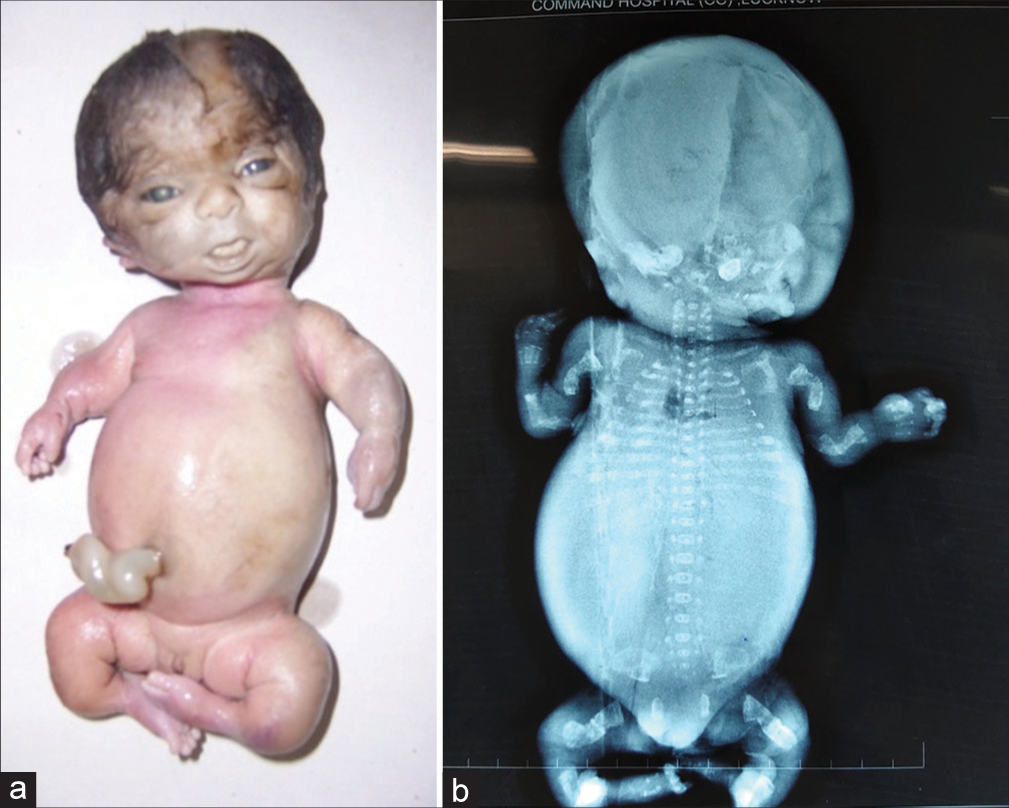
- (a and b) Lethal variety of osteogenesis imperfecta – Type II (a) The fetus showing short and bent limbs, short and narrow thorax (b) Radiograph showing short, bent, crumpled bones (inset), short thorax, beaded ribs and lack of mineralization of skull bones.
Type III variety is also severely deforming and can be picked up in prenatal USG. The children have short bent femora and fractures usually at birth. Though not stillborn, babies with this variety have a risk of death during infancy due to a narrow deformed thorax usually have severe bone deformities, and are usually immobile [Figure 4a and b]. Short stature is severe. Bone density is markedly decreased. The sclera may be blue but becomes normal with age. The face may show flattening with slight prognathism or micrognathia.
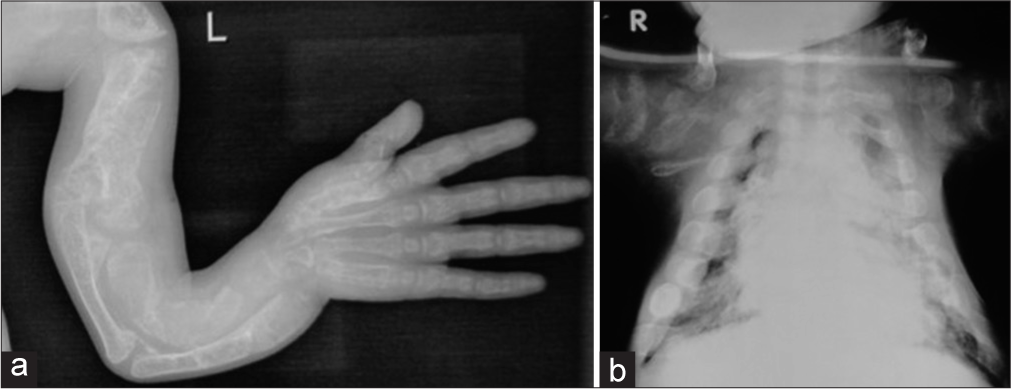
- Radiographs of a child with osteogenesis imperfecta – Type III. (a) Note the deformed bones of the upper limb (b) Narrow thorax.
Type I is a mild form with blue sclerae and hearing loss during adulthood. Height may be normal. OI, type IV is a mild-to-moderate form with normal sclerae. Blue sclerae and dentinogenesis imperfecta are other characteristic features of OI but need not be present in all. Dentinogenesis imperfecta presents as opalescent teeth. The characteristic deep blue sclera is present in type I but absent in type IV. Decreased bone density, age of first fracture, number of fractures, and deformities vary greatly in type I and IV. Height may be normal. Short bent femur with fracture may be seen in a fetus during the third trimester or at birth in OI, types I and IV. Short stature may or may not be present. The round or oval face is seen in some. In addition to the variability of bone deformities, features similar to Ehlers–Danlos syndrome such as joint laxity, easy bruisability, soft skin, and mitral valve prolapse may be present. Some have dilatation of aortic root or aortic regurgitation. Ehlers–Danlos syndrome, type II due to mutations in COL1A2 is a disorder allelic to OI and hence, explains the overlap of phenotypes. Hearing deficits may manifest in teenagers and progress to deafness by midage. Pectus deformities and kyphoscoliosis can be very severe in type III OI. The resultant pulmonary function compromise is an important medical issue and risk factor in some cases.
The radiological evaluation is important for confirmation of diagnosis and documentation of deformities of long bones, abnormalities of spinal curvature, and flattening or compressions of vertebrae. Decreased bone density is usually obvious in radiographs but in mild cases, it may not be very obvious. Good visibility of intracranial structures closer to the USG probe in prenatal USG is a clue to the ossification defect of calvarial bones. The presence of other features such as large anterior fontanelle, wormian bones, basilar invasion, protrusion acetabuli, beaded ribs, hyperplastic callus formation, biconcave appearance of vertebrae (described as codfish vertebrae), and popcorn calcification (the metaphyses and epiphyses exhibit numerous scalloped radiolucent areas with sclerotic margins) helps the diagnosis. Ossification of the interosseous membrane in forearm bones is characteristic of OI and type V [Figure 5].
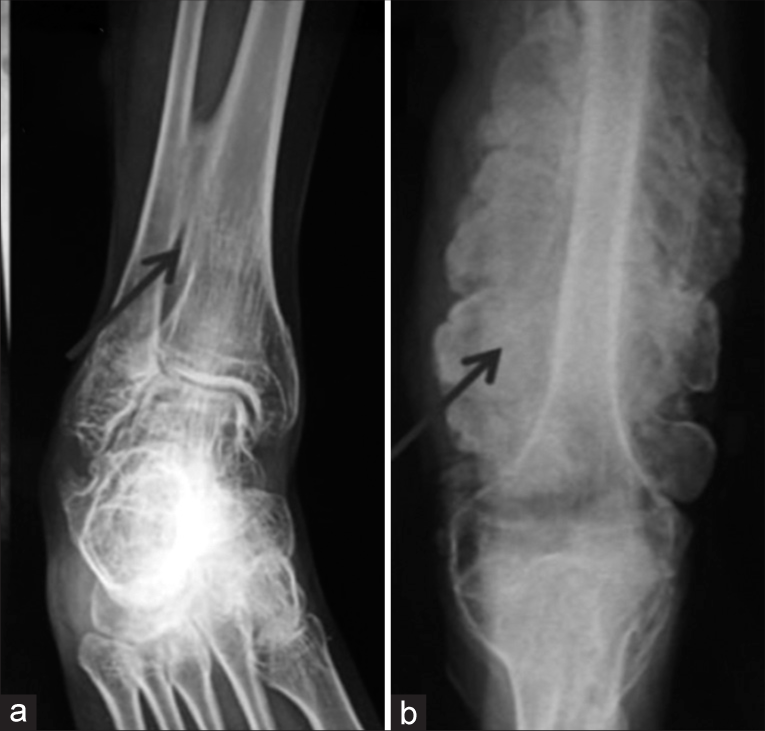
- Radiographs of osteogenesis imperfecta – Type V showing (a) Ossified interosseous membrane (b) Hypertrophied callus.
The clinical and radiological-based diagnosis is usually easy and reliable. However, disorders with decreased bone density may have overlapping features and need to be considered in differential diagnosis. The lethal disorders such as achondrogenesis, hypophosphatasia, and disorders with bent bones such as campomelic dysplasia and kyphomelic dysplasia may be difficult to differentiate from a lethal variety of OI stressing the necessity of DNA-based diagnosis. Postnatally, OI may have phenotypic overlap with cleidocranial dysostosis (large fontanelle and wormian bones), syndromes of osteolysis (Winchester syndrome and Hadju–Cheney syndrome), metabolic bone disorders (renal failure and various types of genetic rickets), osteoporosis pseudoglioma syndrome (pseudoglioma, microphthalmia, and cataract), geroderma ostedysplasticum (pre-mature aging appearance, wrinkled skin, and droopy cheeks), Cole–Carpenter syndrome (craniosynostosis, proptosis, and characteristic facial features) which are associated with decreased bone density. Bruck syndrome is a syndrome of congenital joint contractures and pterygia with decreased bone density and is allelic to OI type XI; both being caused by mutations in the FKBP10 gene. The presence of atypical features should lead to the suspicion of other genetic or non-genetic causes of osteoporosis. Inconsistent history, specific patterns of fractures, unusual family issues, etc., may point toward a battered child or child abuse.
ETIO-PATHOGENIC CLASSIFICATION
Defects in collagen I as a cause of OI were discovered in 1986.[5] Soon it became known that some cases of OI are because of mutations in the COL1A1 gene and some from the COL1A2 gene.[6] Cases of OI with different severities were linked to heterozygous mutations in these two genes. Type I collagen is a trimer comprising two identical α1 (I) polypeptide chains and one α2 (I) polypeptide chain coded by COL1A1 and COL1A2 genes, respectively, and is the major protein of bone matrix (90%). Proper folding of its triple helix is crucial for forming the matrix of bones and other tissues. In collagen, glycine is required at every third position, because the assembly of the triple helix puts this residue at the interior (axis) of the helix. Replacement of the glycine by a bulky amino acid disrupts the triple helix stability and leads to a severe type of OI. This is described as a negative dominant effect. On the other hand, the nonsense mutations causing stop codon leading to prevention of synthesis of procollagen polypeptide does not cause a severe type of OI. It means a decrease in protein synthesis is less harmful than a synthesis of abnormal protein. Hence, all variants in the glycine codon are harmful and pathogenic.
This genotype-phenotype correlation helped in explaining variable phenotypes of OI. Both these genes cause autosomal dominant OI and there were siblings affected with OI, usually type II or III; born to normal parents. This was explained by germline mosaicism. Germline mosaicism has been documented but autosomal recessive genes for OI have been identified and the risk of recurrence in the siblings of such children born to normal parents can be 25%.
The first gene for autosomal recessive OI was identified in 2006.[1] This was the CRTAP gene encoding cartilage-associated protein (CRTAP) and was found to be the cause of OI type VII. At present, in addition to two collagen genes 18 more genes for OI have been identified. These include 16 autosomal recessive genes, one X-linked, and one autosomal dominant gene. Other than COL1A1 and COL1A2, IFTM5 is a cause of an autosomal dominantly inherited OI.[7] A heterozygous gain of (new) function variant in the 5’untranslated region (c.-14C > T) of the IFTM5 gene is associated with a moderate type of OI (OI type V) with distinctive radiographic findings. This pathogenic variant creates a new start codon, resulting in the elongation of the cytoplasmic N-terminus of IFITM5 protein by five amino acids and inducing increased bone formation.[7] The characteristic radiographic findings include hyperplastic callus formation, calcification of the interosseous membrane of the forearm, and hyperdense metaphyseal band. Some patients may present radial head dislocation.
The other genes causing rare types of OI are involved in post-translational modifications, processing, folding, and cross-linking of type I collagen, as well as in bone mineralization and osteoblast differentiation. Hence, OI is now considered to be a disease “associated” with type I collagen. Based on the functions of the protein, a classification has been proposed.[8] The functional classification based on the metabolic mechanism is also proposed by Panzaru et al.:[9]
Group A – Defects in collagen synthesis, structure, or processing
Group B – Defects in collagen modification
Group C – Collagen folding and cross-linking defects
Group D – Compromised bone mineralization
Group E – Defects in osteoblast development with collagen insufficiency.
The formation of pre-type I collagen is a very complex process that requires several post-translational modifications and folding. Proline 3-hydroxylation at position 986 is an important step. The genes for proline hydroxylase complex, namely: propyl 3-hydroxylase 1 (P3H1),[10] CRTAP, and cyclophilin B work together and their functions are interdependent, and the phenotypes of OI resulting from mutations in P3H1 and CRTAP genes are similar. The recent reviews by Yu et al.[11] and Panzaru et al.[9] provide updates on genes causing OI, functions of the proteins encoded by the genes, and classifications based on pathogenic mechanisms. Online Mendelian Inheritance in Man database uses a mixed-genetic classification, with types I–IV according to the Sillence classification and pathogenic variants in COL1A1 or COL1A2, and OI types V to XXII as per the new gene-classified type [Figure 6]. The classifications help in understanding molecular mechanisms and also are useful in clinical practice. [Table 1] describes the mechanisms of various genes causing OI based on the classification by Yu et al.[11]
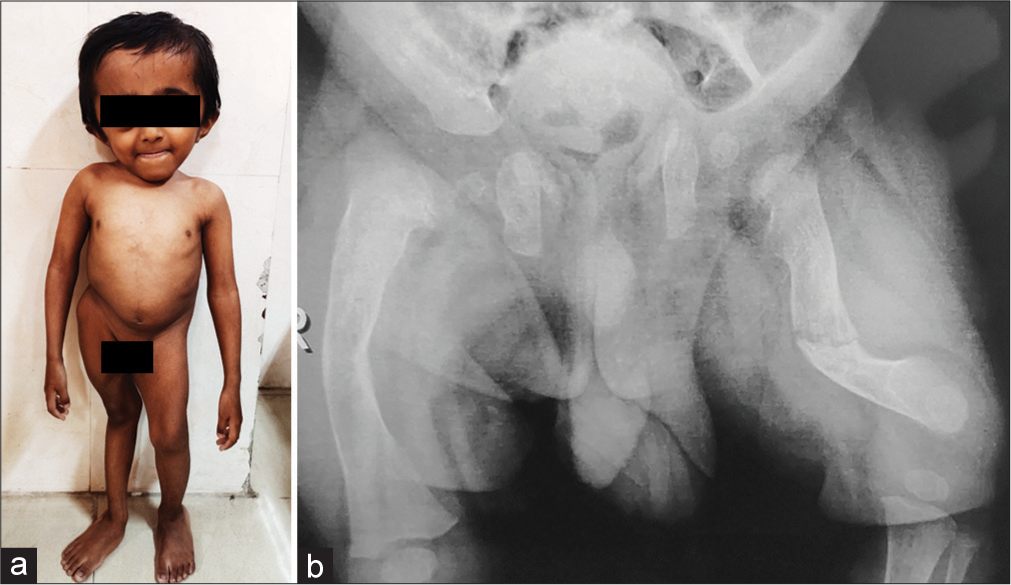
- (a) Eight-year-old boy with osteogenesis imperfecta, type X with causative variant in SERPINH1 gene. (b) His radiograph of pelvis and femora.
| OI group | Mechanism | Type of OI as per OMIM (causative gene) | Comment |
|---|---|---|---|
| Type I A | Collagen synthesis | • Type I (COL1A1) • Types II, III, IV (COL1A1, COL1A2) |
Missense mutations have a more severe phenotype as compared to nonsense mutations. |
| Type I B | Collagen processing | • Type XIII (BMP1) | After normal pre-type I collagen is secreted extracellular, its carboxyl-terminal and amino-terminal properties are cleaved by BMP1 and ADAMT-2, respectively, to form mature type I collagen. |
| Type 1 C | Post-translational modifications | • Type VII (CRTAP) • Type VIII (LEPRE1) • Type IX (PPIB), • Type XIV (TMEMB38) |
The proteins coded by these genes are involved in the folding and proline hydroxylation of the collagen molecules. |
| Type I D | Collagen folding and cross-linking defects | • Type X (SERPINH1) • Type XI (FKBP10) • Type XXI (KDELR2) |
The HSP47 encoded by the SERPINH1is a chaperone protein. The proteins encoded by the FKBP10and PLOD2genes maintain their structural stability and cause lysine hydroxylation of pre-type I collagen telopeptides, respectively. The KEDLRmutation disrupts collagen formation by keeping the HSP47 protein bound to pre-type I collagen. |
| Type II | Bone mineralization defects | • Type V (IFITM5) • Type VI (SERPINF1) |
Defects in SERPINF1increase osteoclast numbers and bone resorption by affecting the OPG/RANKL/RANK pathway, resulting in reduced bone mineralization. IFITM5acts through SERPINF1. |
| Type III | Defective osteoblast differentiation and function | • Type XVI (CREB3L1), • Type XIX (MBTPS2), • Type XV (WNT1), Type XII (SP7), Type XVII (SPRAC) |
WNT1is responsible for osteoblast differentiation and SP7is its target gene. CREB3L1and MBTPS2work together at the osteoblast level. SPRAC(osteonectin) accounts for the unique property of bone collagen to undergo calcification. |
| Type IV | Unclassified | • OI Type XVIII (TENT5A), • OI Type XX (MSED), • OI Type XXII (CCDC134) |
The functional pathways of these proteins are getting understood. |
Types of OI are given as per the Online Inheritance in Man (https://omim.org/) (Based on Yu H, Li C, Wu H, Xia W, Wang Y, Zhao J, et al. Pathogenic mechanisms of osteogenesis imperfecta, evidence for classification. Orphanet J Rare Dis 2023;18:234). OI: Osteogenesis imperfecta, BMP1: Bone morphogenetic protein 1, HSP47: Heat shock protein 47
GENETIC DIAGNOSIS AND MUTATION SPECTRUM
Prenatal or postnatal diagnosis of a typical case is easy and mostly the clinical diagnosis is possible. However, for genetic counseling and prenatal diagnosis, DNA-based diagnosis with the identification of causative variants is essential and stresses the need for molecular diagnosis in each case. Historically for many decades, only COL1A1 and COL1A2 were the only genes known for OI, and collagen analysis followed by sequencing for these genes was the only way for detection of mutations. At present, there are three genes for autosomal dominant OI and 16 genes for autosomal recessive OI. Few families showing X-linked inheritance are due to mutation in MBTPS2. Mutations in the two genes for collagen accounted for 90% of the cases of OI. However, Indian studies showed that 30–50% of cases with OI are because of autosomal recessive genes.[13,14] This is due to high levels of consanguinity and inbreeding. The study from Turkey also showed that autosomal recessive genes account for about 35% of cases with OI.[15]
Genetic heterogeneity and overlap with other genetic and non-genetic causes of osteoporosis pose a challenge. There are hardly any clinical clues to select any one gene for sequencing except ossification of the interosseous membrane and hypertrophic callus formation in type V OI. Most cases of type V have only one causative variant and Sanger sequencing for the variant will be a cheap and easy way. For lack of any such characteristic features; sequencing of all genes for OI and other disorders with osteoporosis should be done by NGS. A panel of genes for OI or whole-exome sequencing (ES) can be ordered.
Accurate three-generation family history in the form of a pedigree and clinical evaluation of family members is essential. The presence of consanguinity suggests the possibility of an autosomal recessive gene. ES of the affected individual/s along with that of affected or unaffected parents in one go is an effective strategy. In case of cost constraints, NGS-based testing of only one affected individual is also useful as the diagnostic yield for OI is very high. Segregation of the variant/s identified in the proband in the family by Sanger sequencing of the parents and siblings is essential for interpretation of the variants of uncertain significance and to know the inheritance of the variant.
The pathogenic variants in COL1A1 and COL1A2 genes in sporadic cases are de novo and will not be present in the parents. The disease-causing variants in COL1A1 and COL1A2 genes usually are missense variations in the codon for glycine or truncating variants leading to the absence of protein synthesis. More than 1,000 pathogenic/likely pathogenic variants are described in the COL1A1 gene and the number is 550 for the COL1A2 gene. Fifty percent of the causative variants in Indian patients are novel[12,13] and guidelines of the American College of Medical Genetics and the American College of Pathologists need to be followed to make reliable interpretations about the pathogenic nature of the variant.[16] In the case of NGS-based testing, sometimes variants in more than one gene may be identified. The laboratory and clinical geneticists need to discuss such variants before finalizing the results. Reevaluation of the patient and relatives for the features of the disorder related to the variant/variants identified; also known as reverse phenotyping may be needed.
OI due to CRTAP, LEPRE1, WNT1, SERPINF1, BMP1, and FKBP10 has been reported from Indian patients in addition to those due to autosomal dominant inheritance.[14,17] A gene-based diagnosis is also required as the standard treatment with BPs is not equally effective in all types of OI.[18] The diagnostic yield of NGS-based testing of OI cases is around 77.6% as reported in a large study of 157 cases from Brazil.[18] The reasons for not identifying genetic variations in some cases may be various. ES does not identify deep intronic variations, some copy number variations, and variations in gene-controlling regions. Further testing by whole-genome sequencing and new algorithms for analysis may identify variations in some more cases. Moreover, some genes for OI may be still unknown to us and these cases will need to be further studied by scientists.
The phenotypes are extremely variable as are genotypes. The missense variations in collagen genes are more harmful than the truncating variants. However, there are no significant genotype-phenotype correlations. For autosomal dominant genes, the penetrance is complete though some family members carrying the pathogenic variant may be very mildly affected. Type II (perinatally lethal) and type III (severe deforming) are severe types and can be due to recessive or dominant genes. Many of these cases present prenatally even before 20 weeks of gestation and are detected in routine prenatal USG in families without any case of OI. Some milder cases also may present during the third trimester with short bent femora. ES of such fetuses, prenatally or postnatally is essential for genetic diagnosis. Based on the genetic diagnosis, accurate risk of recurrence and prenatal diagnosis can be provided; if the family wishes to avoid the birth of a similarly affected child in the future. About 20% of fetuses with short long bones are OI.[20]
MANAGEMENT
Out of many drugs tried, BPs (intravenous as well as oral) are found to be effective in improving outcomes. Although many studies show a decrease in fracture rates with BP therapy, the efficacy varies greatly.[21] Though the frequency of fractures may not reduce in some patients; the patients report decrease in pain and improvement in the quality of life. Bajpai et al. have reported their experience in Indian patients with 3 years of experience that bone density increases and fracture rates decrease.[22] Zoledronic acid also has been used in children and found to be safe, thus allowing 6 monthly dosings.[23] The duration of therapy and whether to stop BPs before surgery are the two questions that are still under research. Maintenance of adequate levels of vitamin D should be ensured.[24] Surgical interventions need to be done at specialized centers as the techniques and decisions on when to interfere is a very specialized area of expertise.
BPs are used regularly for most of the patients and have changed the outcomes for many; still more effective drugs are needed. The efficacy may be dependent on the genetic etiology. Understanding the roles of various proteins other than collagen in bone ossification and mineralization will lead to the development of novel drugs specific to different types of OI. The role of denosumab which prevents interaction between RANKL and RANK was considered a strong drug candidate for OI patients, particularly for patients with the BP-unresponsive types. The evidence for denosumab’s effects in children with OI indicates that it effectively improves bone mineral density but not fracture rates.[25] Anti-sclerostin agents and transforming growth factor-beta antagonists are under investigation in clinical trials.[26] Gene therapy for OI is also under research.[28]
The education of children and families and psychological support are an important part of management. The children need to live with the disease which continues to interfere in the day to day life activities and may be associated with handicaps and affect longevity. Many role models conquering the disease are available and their involvement in patient support groups will help others to inculcate a positive attitude toward life.[29]
GENETIC COUNSELING AND PRENATAL DIAGNOSIS
OI is a phenotype associated with collagen and extreme genetic and phenotypic heterogeneity makes genetic counseling a very complex process. The mode of inheritance can be autosomal dominant, recessive, or X-linked. Positive family history may be present in some but many cases are sporadic. Recurrence in sibs can be due to autosomal recessive mode of inheritance or due to germline mosaicism. Parents may have mild manifestations. Keller et al. have reported a family where the heterozygous parents were mildly affected while the homozygous offspring was severely affected with the phenotype of type II, lethal variety.[30] Many of these issues can be addressed and accurate genetic counseling can be provided if a genetic diagnosis of the proband is done and causative variant/variants are detected in the proband (affected individual in the family).
Autosomal recessive types of OI are usually severe and the risk of recurrence is 25%. The families may opt for prenatal diagnosis in such cases. DNA-based prenatal diagnosis is more reliable and can be done at 12 weeks of gestation. USG-based diagnosis of types II and III may be possible before 20–24 weeks of gestation.
For families with autosomal dominant varieties of OI, if one of the parents is affected, the risk in the offspring is 50%. However, if the parents of a child with OI due to the autosomal dominant gene are normal and not carriers of the genetic variant, the risk of recurrence is not significantly increased. However, recurrence due to germline mosaicism is possible and hence prenatal testing should be offered. Depending on the severity of the proband in the family, they may opt for a prenatal diagnosis. Milder types of OI may not justify prenatal diagnosis. The decision of prenatal diagnosis and termination of the fetus with OI has to be taken by the family and the genetic counseling should be non-directive.
Nowadays, pre-implantation diagnosis and non-invasive diagnosis from free fetal DNA in maternal plasma are other alternatives to prenatal diagnosis based on fetal sampling. These options need counseling by the clinician with a good understanding of these options. Rarely, the options of gamete donations may need to be discussed, especially if the molecular testing has failed to identify genetic etiology.
CONCLUSION
Genetics of OI has evolved and the heterogeneity of genotypes and phenotypes has added many dimensions to the genetic diagnosis and counseling. Genetic diagnosis of each affected child is essential and should be done by NGS-based testing. Genetic diagnosis can provide information regarding outcome and the possibility of recurrence in the family. Limited efficacy of BPs has been realized and a better understanding of the pathology of the disorder may provide new drugs specific to each OI type. The identification of many non-collagen-related genes for OI has strengthened the knowledge of bone density and mineralization and the management in the future will not be limited to genetic counseling but we hope for gene-based therapies or drugs modifying the disease processes at molecular levels.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The author confirms that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- CRTAP is required for prolyl 3-hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291-304.
- [CrossRef] [PubMed] [Google Scholar]
- Phenotyping and genotyping of skeletal dysplasias: Evolution of a center and a decade of experience in India. Bone. 2019;120:204-11.
- [CrossRef] [PubMed] [Google Scholar]
- Analysis of short stature cases referred for genetic evaluation. Indian J Pediatr. 2012;79:1597-600.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic heterogeneity in osteogenesis imperfecta. J Med Genet. 1979;16:101-16.
- [CrossRef] [PubMed] [Google Scholar]
- Osteogenesis imperfecta is linked to both Type I collagen structural genes. Lancet. 1986;2:69-72.
- [CrossRef] [PubMed] [Google Scholar]
- Consistent linkage of dominantly inherited osteogenesis imperfecta to the Type I collagen loci: COL1A1 and COL1A2. Am J Hum Genet. 1990;46:293-307.
- [Google Scholar]
- A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta Type V. Am J Hum Genet. 2012;91:343-8.
- [CrossRef] [PubMed] [Google Scholar]
- Osteogenesis imperfecta due to mutations in non-collagenous genes: Lessons in the biology of bone formation. Curr Opin Pediatr. 2014;26:500-7.
- [CrossRef] [PubMed] [Google Scholar]
- Classification of osteogenesis imperfecta: Importance for prophylaxis and genetic counseling. World J Clin Cases. 2023;11:2604-20.
- [CrossRef] [PubMed] [Google Scholar]
- Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet. 2007;39:359-65.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenic mechanisms of osteogenesis imperfecta, evidence for classification. Orphanet J Rare Dis. 2023;18:234.
- [CrossRef] [PubMed] [Google Scholar]
- Mutation spectrum of COL1A1 and COL1A2 genes in Indian patients with osteogenesis imperfecta. Am J Med Genet A. 2014;164A:1482-9.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone. 2018;110:368-77.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. Eur J Med Genet. 2015;58:21-7.
- [CrossRef] [PubMed] [Google Scholar]
- Osteogenesis imperfecta in 140 Turkish families: Molecular spectrum and, comparison of long-term clinical outcome of those with COL1A1/A2 and biallelic variants. Bone. 2022;155:116293.
- [CrossRef] [PubMed] [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [CrossRef] [PubMed] [Google Scholar]
- Worsening of callus hyperplasia after bisphosphonate treatment in Type V osteogenesis imperfecta. Indian Pediatr. 2016;53:250-2.
- [CrossRef] [PubMed] [Google Scholar]
- Osteogenesis imperfecta Type VI in childhood and adolescence: Effects of cyclical intravenous pamidronate treatment. Bone. 2007;40:638-44.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic analysis of osteogenesis imperfecta in a large Brazilian cohort. Bone. 2023;169:116683.
- [CrossRef] [PubMed] [Google Scholar]
- Should we offer prenatal exome sequencing for intrauterine growth restriction or short long bones? A systematic review and meta-analysis. Am J Obstet Gynecol. 2023;228:409-17.e4.
- [CrossRef] [PubMed] [Google Scholar]
- Bisphosphonates for the prevention of fractures in osteogenesis imperfecta: Meta-analysis of placebo-controlled trials. J Bone Miner Res. 2015;30:929-33.
- [CrossRef] [PubMed] [Google Scholar]
- Intravenous pamidronate therapy in osteogenesis imperfecta: Response to treatment and factors influencing outcome. J Pediatr Orthop. 2007;27:225-7.
- [CrossRef] [PubMed] [Google Scholar]
- Zoledronic acid in children with osteogenesis imperfecta and Bruck syndrome: A 2-year prospective observational study. Osteoporos Int. 2016;27:81-92.
- [CrossRef] [PubMed] [Google Scholar]
- Vitamin D and osteogenesis imperfecta in pediatrics. Pharmaceuticals (Basel). 2023;16:690.
- [CrossRef] [PubMed] [Google Scholar]
- Denosumab use in osteogenesis imperfecta: An update on therapeutic approaches. Ann Pediatr Endocrinol Metab. 2023;28:98-106.
- [CrossRef] [PubMed] [Google Scholar]
- Approach to the patient: Pharmacological therapies for fracture risk reduction in adults with osteogenesis imperfecta. J Clin Endocrinol Metab. 2023;108:1787-96.
- [CrossRef] [PubMed] [Google Scholar]
- Gene-repaired iPS cells as novel approach for patient with osteogenesis imperfecta. Front Bioeng Biotechnol. 2023;11:1205122.
- [CrossRef] [PubMed] [Google Scholar]
- Multimodal pain management of children diagnosed with osteogenesis imperfecta: An integrative literature review. Pain Manag Nurs. 2023;24:102-10.
- [CrossRef] [PubMed] [Google Scholar]
- 'JAYA' of mind over bones! Genetic clinics. . 2019;12:2-3. Available from: https://iamg.in/genetic_clinics/april_june_2019_heartoheartalk.php [Last accessed on 2023 Oct 02]
- [Google Scholar]
- Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively. Genet Med. 2018;20:411-9.
- [CrossRef] [PubMed] [Google Scholar]




