
Translate this page into:
A case of McCune–Albright syndrome hiding in the bones


*Corresponding author: Gabrielle Doré-Brabant, Department of Pediatrics, CHU de Québec - Laval University, Québec, Canada. gabrielle.dore-brabant.1@ulaval.ca
-
Received: ,
Accepted: ,
How to cite this article: Doré-Brabant G, Rousseau-Nepton I. A case of McCune–Albright syndrome hiding in the bones. J Pediatr Endocrinol Diabetes 2022;2:81-5.
Abstract
McCune–Albright syndrome (MAS) is a rare genetic disorder which presents across a broad clinical spectrum. The characteristic features are café-au-lait macules, fibrous dysplasia of the bone (FD), and hyperfunctioning endocrinopathies. A 10-year-old girl presented with peripheral precocious puberty, growth hormone (GH) excess, and asymptomatic FD without café-au-lait macules. MAS should be suspected in the presence of any characteristic feature, even in the absence of café-au-lait macules. A skeletal survey should be considered in patients presenting with an unknown cause of peripheral precocious puberty to search for signs of FD, even in the absence of other clinical manifestations.
Keywords
McCune–Albright syndrome
Fibrous dysplasia of bone
Precocious puberty

INTRODUCTION
McCune–Albright syndrome (MAS) is a rare genetic disorder with a prevalence estimated to be between 1 in 100,000 and 1 in 1,000,000.[1] It is caused by a post-zygotic somatic gain-of-function mutation in the GNAS gene. This mutation in the GNAS gene results in an active α-subunit of the Gs protein, leading to constitutive receptor activation and inappropriate intracellular cyclic AMP production. This α-subunit portion of the Gs protein is found in multiple organs, including endocrine tissues, bone, and skin. Somatic mutations, which occur early in embryogenesis, are distributed in a mosaic pattern and the clinical presentation is largely determined by the extent and location of mutation bearing tissues. Consequently, MAS presents along a broad clinical spectrum.[2,3]
The original description of MAS was a triad of café-au-lait macules, precocious puberty, and fibrous dysplasia of the bone (FD).[2] The characteristic features now include not only all the initial characteristics in the triad, but also hyperfunctioning endocrinopathies, including gonadotropin-independent gonadal function, growth hormone (GH) excess with acromegaly/ gigantism, non-autoimmune hyperthyroidism, and neonatal hypercortisolism.[3]
The diagnosis is often made clinically, based on the presence of two or more characteristic features.[2,3] Detection of the GNAS mutation is variable depending on the mosaicism of the tested tissue. Mutation detection rates in affected tissue are over 80% in comparison to peripheral blood lymphocytes, which are around 20–30%.[2] Consequently, a negative result does not rule out MAS and detection of the GNAS mutation is not mandatory to establish the diagnosis.
The case of a 10-year-old girl with a diagnostically challenging presentation of MAS is presented below.
CASE REPORT
A previously healthy 5½-year-old girl was referred to the endocrinology outpatient clinic with signs of precocious puberty. Progressive thelarche and pubarche occurred right after her fifth birthday. Physical examination revealed height and weight around the 50th percentile and Tanner stage 2 for breast and pubic hair. No café-au-lait macule, mucosal pigmentation, or skeletal anomaly were visible. There was no previous history of fractures.
Laboratory values [Table 1] were normal except for the estradiol level, with a detectable value on two consecutive samples of 101 and 100 pmol/L. Two luteinizing hormone-releasing hormone (LHRH) stimulation tests showed a prepubertal luteinizing hormone (LH) response (LH peak < 5 U/L and peak LH/follicle-stimulating hormone (FSH) ratio < 0.66).[4] Bone age was not advanced. Pelvic ultrasound revealed a pubertal uterus (1.4 × 2.2 × 5.2 cm) with visible endometrium and ovaries (1.5 mL and 2.7 mL) without ovarian masses. It should be noted that a week before the ultrasound, at the age of 5 years and 9 months, she presented with a 3-day history of moderate periumbilical abdominal pain. The day after the pain resolved she had light vaginal bleeding. A ruptured ovarian cyst, not seen on the ultrasound, was hypothesized at this point.
| Age | 55/12 | 59/12 | 61/12 | 63/12 | 65/12 | 72/12 | 80/12 | 84/12 | 88/12 | 89/12 |
|---|---|---|---|---|---|---|---|---|---|---|
| LH, UI/L | <0.2 | <0.2 | <0.2 | <0.2 | <0.2 | 0.4 | ||||
| FSH, UI/L | 0.9 | <0.3 | 1.3 | 0.4 | <0.2 | 0.6 | ||||
| Estradiol, pmol/L | 101 | 100 | <40 | <40 | 74 | 92 | 73 | <40 | ||
| TSH, mUI/L | 1.56 | 1.17 | 0.71 | 1.05 | 0.04 | <0.02 | ||||
| N=0.25–5.00 | ||||||||||
| FT4, pmol/L | 10.2 | 8.0a | 11.3a | 13.8 | 17.1 | |||||
| N=10.0–19.0 | ||||||||||
| aN=12.0–22.0 | ||||||||||
| Prolactin, µg/L | 4 | 56 | 11 | |||||||
| N<23 | ||||||||||
| IGF1, µg/L | 259 | 307 | 165 | |||||||
| N=80–235 | ||||||||||
| GH, µg/L | 5.9 | |||||||||
| LHRH stimulation test, U/L | ||||||||||
| LH at beginning | <0.2 | <0.2 | <0.2 | <0.2 | 0.4 | <0.2 | ||||
| FSH at beginning | 1.5 | <0.2 | 0.9 | 0.8 | 0.2 | 0.2 | ||||
| Peak LH | 4.5 | <0.2 | 2.7 | 3.8 | 0.9 | 0.3 | ||||
| Peak FSH | 15.6 | 0.8 | 10.3 | 8.8 | 0.3 | 0.3 | ||||
| OGTT, ng/mL | ||||||||||
| Peak GH | 12.5 | |||||||||
| Nadir GH | 0.6 | |||||||||
| Bone age | 59/12 | 710/12 | 710/12 | 710/12 | 100/12 | 110/12 | ||||
| Chronologic age | 56/12 | 62/12 | 66/12 | 72/12 | 80/12 | 88/12 |
LH: Luteinizing hormone, LHRH: Luteinizing hormone-releasing hormone, FSH: Follicle-stimulating hormone, TSH: Thyroid-stimulating hormone, FT4: Free thyroxine, IGF1: Insulin-like growth factor 1, GH: Growth hormone, OGTT: Oral glucose tolerance test. Bolded values are those outside the normal range. Underlined values are those obtained under treatment
Between the ages of 6 years and 7 years, her growth accelerated from the 55th (+0.14 SD) to the 86th (+1.10 SD) percentile with a growth velocity of 15 cm/year [Figure 1]. She also had five irregular menstrual cycles with bleeding without preceding abdominal pain during this period. Laboratory values [Table 1] were all normal, including an undetectable estradiol. Two additional LHRH stimulation tests also showed a prepubertal LH response. Bone age was done on three separate occasions and showed progression to 710/12 years. Pelvic ultrasound was comparable to the previous one, except for an increase in the volume of the ovaries (24 mL and 30 mL). Due to the pubarche accompanying the thelarche and because the result of the LHRH stimulation test was not typical of a peripheral origin, central precocious puberty with fluctuating symptoms was hypothesized. Pituitary MRI with gadolinium and dynamic study was normal.
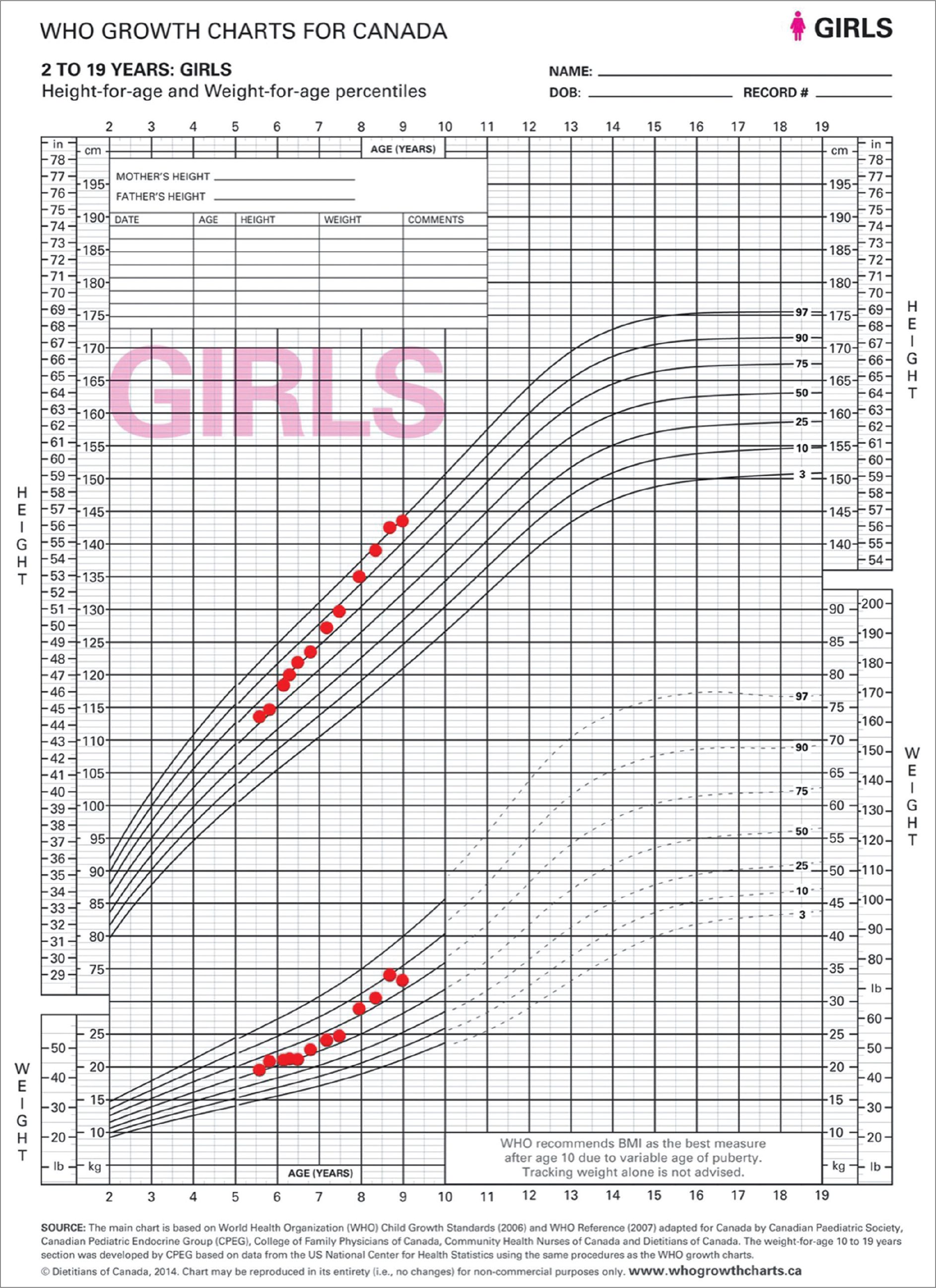
- A 10-year-old girl with signs of precocious puberty and growth acceleration secondary to McCune–Albright syndrome. Growth curve from the first endocrinologist outpatient clinic visit to the diagnosis.
Between the ages of 7½ and 8 years, she had her menses every month, her growth velocity accelerated with a gain of 5.1 cm in 6 months, her height rose to the 92nd percentile (+1.41 SD), and her Tanner stage progressed to B3P2A1. Laboratory values [Table 1] were normal, except for a detectable estradiol level of 92 pmol/L, and a mildly decreased free thyroxine (FT4) with a normal thyroid-stimulating hormone (TSH) level. Bone age was advanced to 100/12 years. Given the acceleration in growth velocity and the advancement in bone age in the context of a diagnosis of central precocious puberty, leuprolide 7.5 mg IM monthly was started at the age of 8 years.
Four months after the start of leuprolide, she did not have any other signs of puberty except her growth velocity, which was 4.2 cm in 4 months, increasing her height to the 96th percentile (+1.77 SD). Somewhat coarse facial features and rare complaints of excessive sweating were noted. A repeat LHRH stimulation test continued to show suppression of the axis. The low FT4 was persisting over three samples, despite the absence of clinical signs of hypothyroidism. Levothyroxine was started to treat central hypothyroidism. Hypopituitarism screening revealed a normal morning cortisol (221 nmol/L) and mildly elevated insulin-like growth factor 1 (IGF1) and prolactin levels [Table 1].
At the age of 8 years–8 months, she had menses on 3 out of 4 months with periumbilical abdominal pain preceding each menses. Her Tanner stage progressed to B3P3A1. Laboratory values [Table 1], including estradiol and prolactin levels, were normal, except for IGF1 which was still elevated. Bone age advanced to 110/12 years. LHRH stimulation test showed again good suppression of the axis. A 3-h oral glucose tolerance test was performed in the context of the elevated IGF1 and growth velocity acceleration. This was diagnostic for GH excess with a paradoxical GH peak of 12.5 ng/mL at 60 min and a nadir of 0.6 ng/mL. Of note, the detection level of GH is <0.1 ng/mL.[5,6] A genetic panel of gigantism genes, including GNAS mutation, identified no clinically significant sequence or variants in peripheral blood. A subsequent MRI of the pituitary gland without gadolinium was normal. Given the lack of pubertal suppression on leuprolide, peripheral precocious puberty was diagnosed. A repeat pelvic ultrasound showed a left ovarian cyst of 2.5 × 4.8 cm. Subsequently, a bone scintigraphy revealed multiple fibrodysplasic lesions [Figure 2], especially in the posterior occipital and fronto-zygomatic bones, as well as in the right humerus, shin bones and left femur.
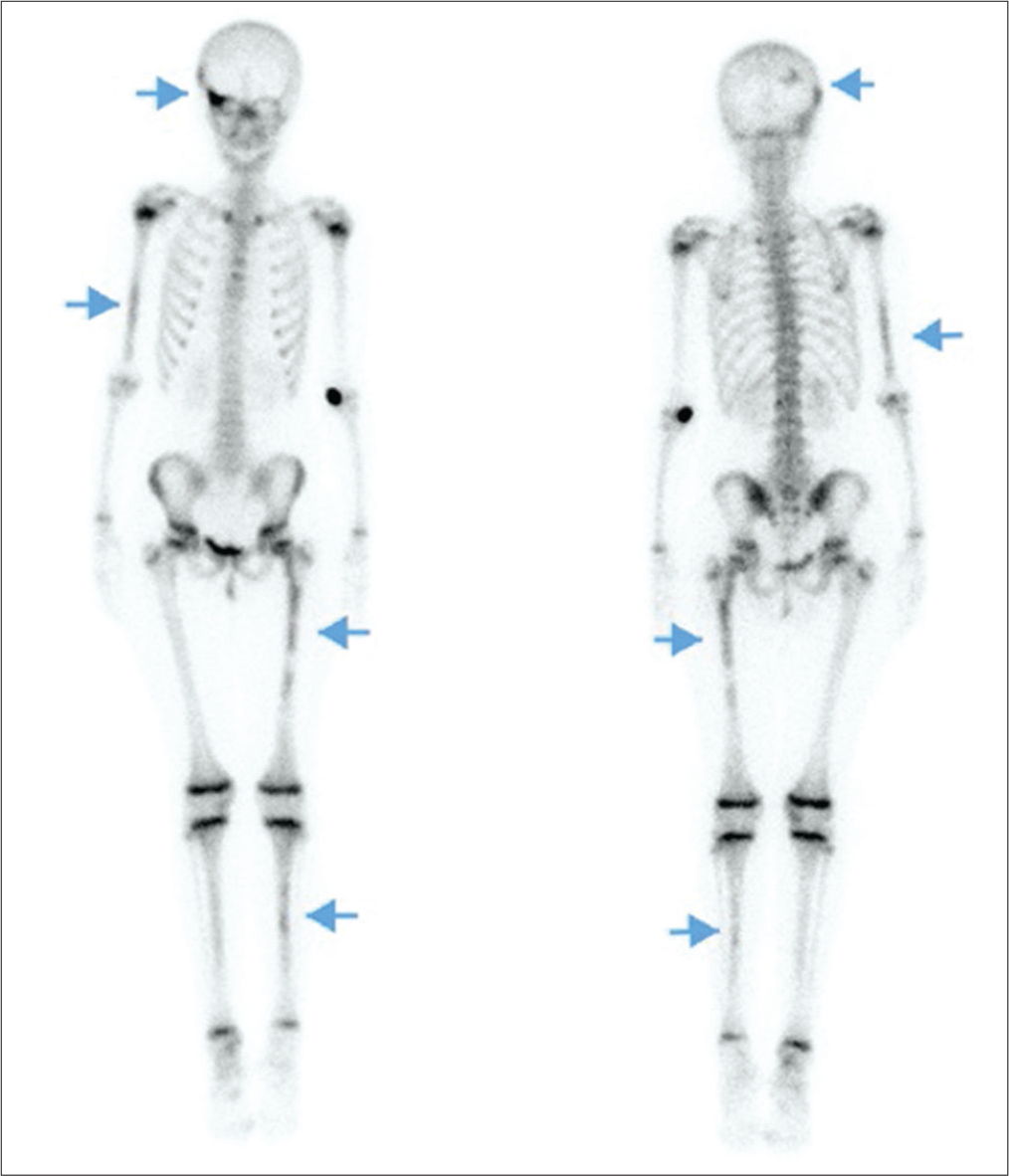
- A 10-year-old girl with McCune–Albright syndrome without bone pain nor skeletal anomaly. Bone scintigraphy showing fibrodysplasic lesions (arrows) in the right posterolateral occiput and the upper part of the right orbit in the frontozygomatic bones, as well as in the distal two-thirds of the right humerus, the proximal two-thirds of left femur and a few foci in shin bones.
With the presence of three characteristic features, namely, peripheral precocious puberty, gigantism, and FD, the clinical diagnosis of MAS was made. Letrozole 2.5 mg PO daily and octreotide 50 mg SQ 3 times daily were started, and leuprolide was continued. Octreotide was switched to 10 mg IM/month after 1 month since GH was not suppressed. Phosphorus, alanine transaminase, lipase, glucose, and fasting lipid profile were within normal limits. Ophthalmologic examination revealed no evidence of optic neuropathy. Orthopedic examination revealed leg length discrepancy with the left limb being one centimeter shorter.
DISCUSSION
The diagnosis of MAS was based on the cumulative signs arising from tissues affected by the GNAS mutation. Since the presentation is variable, MAS can be challenging to diagnose.
Moreover, establishing the peripheral origin of puberty and the diagnosis of gigantism can be difficult.
First, this case emphasizes that even if estradiol is undetectable, both etiologies of puberty need to be considered.[7] In peripheral precocious puberty, estradiol is often detectable with suppressed LH and FSH. In this case, estradiol was often undetectable despite the peripheral origin. When detectable, levels of estradiol were likely explained by the presence of an ovarian cyst or follicle. Ovulatory follicle resolution led to a decline in estradiol levels down to a prepubertal value.[3] In this patient, the presence of cysts was sometimes accompanied by abdominal pain and by menses in the next days, but most episodes were asymptomatic except for the vaginal bleeding. The first two ultrasound scans were done days after menses, which probably explains why no cysts were seen on those studies. Due to the pubarche accompanying the thelarche, central precocious puberty was hypothesized, although the irregular menses with the normal LHRH stimulation tests were pointing toward a peripheral origin of the puberty. The pubarche was likely due to premature adrenarche and not related to MAS. This case emphasizes that the origin of precocious puberty cannot be based on estrogen levels and that a normal ultrasound does not rule out a peripheral cause.
Second, FD more often manifests with pathologic fractures, bone pain, painless facial asymmetry, or skeletal anomalies in MAS.[2] Our patient was completely asymptomatic as described by Kelly et al. in children with FD.[8] Early identification of FD allows prompt detection of complications related to its location (deafness, vision loss, cranial nerves palsy, etc.).[2] Furthermore, this case highlights that patients presenting with an unclear cause of peripheral precocious puberty without other features of MAS could benefit from a skeletal survey, searching for evidence of FD, even in the absence of its clinical manifestations.
Third, pituitary involvement, with GH excess in this case, is uncommon in MAS, occurring only in 10–15% of patients.[3] For the affected patients, 85% of them have both GH and prolactin excess, which were not the case for our patient.[3] In this case, the GH excess was subtle, but diagnostic with the 3-h oral glucose tolerance test. Surprisingly, for this patient, GH excess did not cause any FD symptoms, even though imaging shows multiple FD lesions, including some in the craniofacial region (which is usually affected by GH excess).[3] In relation to GH excess, we hypothesize that the low FT4 was related to the increasing peripheral conversion of thyroxine (T4) to triiodothyronine (T3) without clinical symptoms of hypothyroidism in this patient.[9] A low FT4 with a normal TSH could be interpreted as central hypothyroidism, which complicated the diagnostic picture.
CONCLUSION
We present a peculiar case of MAS presenting with a challenging diagnosis of peripheral precocious puberty associated with asymptomatic FD and GH excess resulting in accelerated metabolism of FT4, mimicking central hypothyroidism, in a patient without café-au-lait macules.
Learning points
Central versus peripheral precocious puberty cannot be distinguished by estrogen levels.
Normal pelvic ultrasound, without ovarian cyst, does not rule out peripheral causes of precocious puberty.
A skeletal survey, searching for asymptomatic FD, should be considered in patients presenting with an unclear cause of precocious puberty.
Suspect MAS in the presence of characteristic features, even in the absence of café-au-lait macules.
Acknowledgment
We would like to thank Dr. Geneviève April for providing the bone scan image and Dr. Mary Jiang for the English revision of the manuscript.
Authors’ contributions
All the authors have contributed equally to the manuscript.
Ethical approval
Not applicable.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- The clinical spectrum of McCune-albright syndrome and its management. Horm Res Paediatr. 2019;92:347-56.
- [CrossRef] [PubMed] [Google Scholar]
- Fibrous dysplasia/McCune-albright syndrome: A rare, mosaic disease of Gα s activation. Endocr Rev. 2020;41:345-70.
- [CrossRef] [PubMed] [Google Scholar]
- Gonadotropin-releasing hormone stimulation test and diagnostic cutoff in precocious puberty: A mini review. Ann Pediatr Endocrinol Metab. 2020;25:152-5.
- [CrossRef] [PubMed] [Google Scholar]
- The biochemical diagnosis of acromegaly. J Clin Med. 2021;10:1147.
- [CrossRef] [PubMed] [Google Scholar]
- Growth hormone response to oral glucose load: From normal to pathological conditions. Neuroendocrinology. 2019;108:244-55.
- [CrossRef] [PubMed] [Google Scholar]
- Estradiol levels in prepubertal boys and girls-analytical challenges. Int J Androl. 2004;27:266-73.
- [CrossRef] [PubMed] [Google Scholar]
- Pain in fibrous dysplasia of bone: Age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19:57-63.
- [CrossRef] [PubMed] [Google Scholar]
- The interrelationships of thyroid and growth hormones: Effect of growth hormone releasing hormone in hypo-and hyperthyroid male rats. Acta Endocrinol Suppl (Copenh). 1986;279:367-75.
- [CrossRef] [PubMed] [Google Scholar]




