
Translate this page into:
Refractory rickets: A case of X-linked hypophosphatemic rickets (PHEX gene variation)

*Corresponding author: Dhanya Soodhana Mohan, Department of Pediatric and Adolescent Endocrinology, Aster Malabar Institute of Medical Sciences, Calicut, Kerala, India. dhanyasoodhana@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mohan DS, Veettil RT, Vijayakumar M. Refractory rickets: A case of X-linked hypophosphatemic rickets (PHEX gene variation). J Pediatr Endocrinol Diabetes. 2024;4:35-9. doi: 10.25259/JPED_43_2023
Abstract
Hypophosphatemic rickets is a type of hereditary rickets distinguished by both hypophosphatemia and hyperphosphaturia. X-linked hypophosphatemic rickets (XLH) is the most prevalent type of heritable hypophosphatemic rickets. We describe a 3-year-old child whose phosphate-regulating endopeptidase homolog X-linked (PHEX) gene variation resulted in lower limb abnormalities and short stature. A similar history was noted in the father. We highlight the significance of a prompt diagnosis and initiation of treatment to prevent subsequent sequelae.
Keywords
Rickets
Hypophosphatemia
PHEX gene

INTRODUCTION
Rickets is a disorder of the growing child that occurs due to the deferred mineralization of the growth plate due to impaired apoptosis of the growth plate cartilage.[1] Lack of calcium (Ca) and vitamin D due to the underlying malnutrition is the most common cause of rickets among children in India. Refractory rickets need to be considered if the rickets fails to heal or a family history of rickets is present.[2] Refractory rickets is frequently caused by hypophosphatemic rickets, renal tubular acidosis, Fanconi syndrome, vitamin D-dependent rickets (VDDR), chronic renal failure, and malabsorption. One of the main types of refractory rickets is hereditary hypophosphatemic rickets, which can be brought on by mutations in the PHEX, FGF23, DMP1, ENPP1, and SLC34A3 genes.[3] The most common form of phosphopenic rickets is XLH, occurring in about 1 in 20,000 newborns. Over 200 distinct variants in the PHEX gene have been found, and inactive mutations in the gene, which is found in Xp22.1-22.2, have been linked to the pathophysiology of XLH.[4]
Chronic hypophosphatemia is the denominator of all forms of rickets. Low phosphorus levels in the extracellular space cause the hypertrophic chondrocytes to fail at apoptosis, which impairs the growth plate’s mineralization. Parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF23) play a crucial role in preserving normophosphatemia by facilitating the renal excretion of phosphorus by acting on the proximal renal tubules. Nutritional disorders and VDDR lead to persistently low phosphorus levels due to an increase in PTH levels and lead to rickets.[2]
We present a case of rickets where a detailed family history provided us with a clue to the probable genetic etiology of rickets.
CASE REPORT
A 3-year-old girl was referred to the pediatric endocrinology clinic of our hospital (a tertiary care center) as a case of resistant rickets. She had presented to a pediatrician with complaints of difficulty in walking since the age of 1.5 years and bowing of legs noted since the age of 2 years. She did not complain of muscle weakness or bony pain. She consumed a predominantly meat-based diet and drank two glasses of milk daily. She also played outdoors every day. She did not have polyuria and was gaining weight well, according to the mother. She had no history of fractures or dental abscesses. On examination, she was active with stable vitals. Her weight was 8.48 kg (–4.11 SDS), her height was 75 cm (–5.27 SDS), and she was within the target height. The father’s height was 137 cm and weight 59 kg; the mother’s height was 154 cm and weight 48 kg. The calculated target height was 139 cm, which was below the 3rd centile. On general physical examination, she had frontal bossing and mild widening of the wrists; rachitic rosary was present but very subtle and had genu valgus deformity of the right lower limb [Figure 1]. There was anterior bowing of the right lower limb, too. Systemic examination was normal.
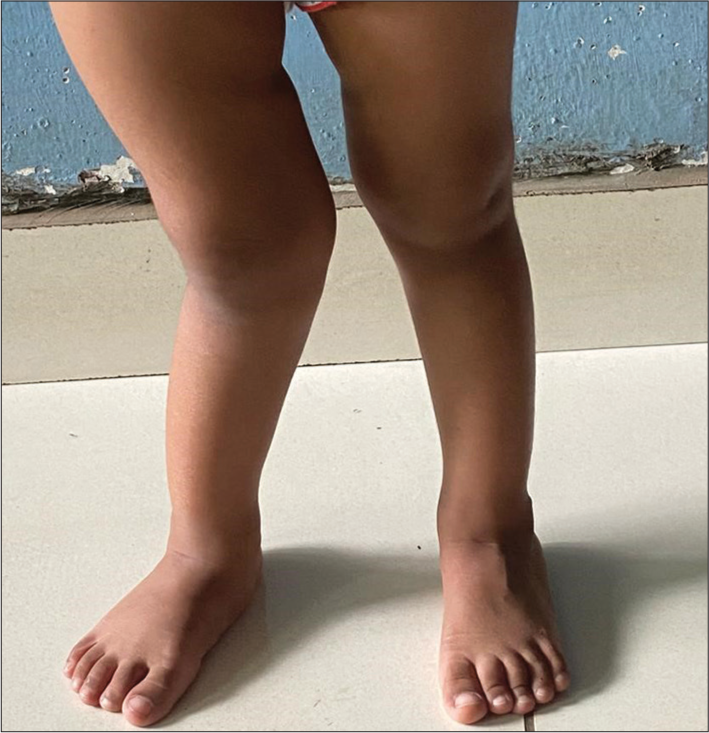
- Genu valgus deformity of the right lower limb.
Initial blood investigations showed serum Ca 9.1 mg/dL (normal range 9.4–10.8 mg/dL), serum phosphate 3 mg/dL (normal range 3.8–6.5 mg/dL), alkaline phosphatase (ALP) 1598 IU/L (normal range 265–849 U/L), 25-hydroxyvitamin D level of 29 ng/mL (normal range 30–100 ng/mL), and PTH level was 173 pg/mL (normal range 10–55 pg/mL). The radiograph of the hand and wrist showed widening and fraying of the metaphyseal plate, coarse trabeculations, and thick cortices [Figure 2].
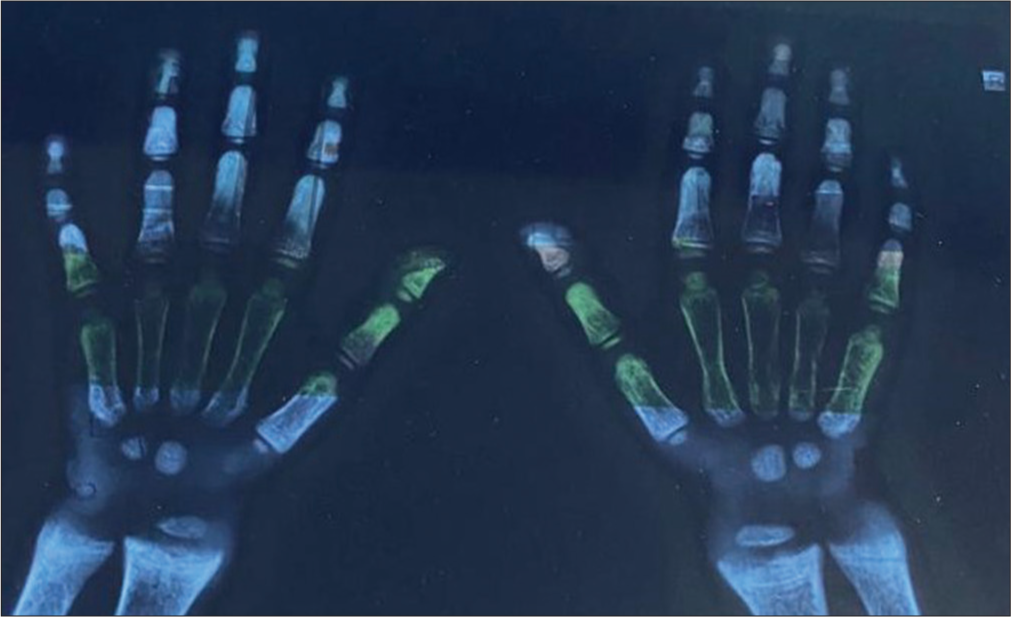
- X-ray of the hand and wrist showing widening and fraying of the metaphyseal plate, coarse trabeculations, and thick cortices, which are prominent in X-linked hypophosphatemic rickets.
Given the clinical features of rickets, she was given vitamin D 60,000 IU once a week for six weeks and oral Ca 70 mg/kg/day for two months by the treating pediatrician, and on follow-up serum phosphate continued to be low (3.2 mg/dL), and ALP level had not come down (2104 IU/L). On further evaluation, 25-hydroxyvitamin D was high (>150 ng/mL), and serum phosphate was persistently low at 2.8 mg/dL. The arterial blood gas analysis was normal (pH 7.39, pCO2 38.2 kPA, HCO3 22.7 mmol/L); serum creatinine was 0.3 mg/dL; and urine examination was normal. 24-hour urine Ca to creatinine ratio and ultrasound of the abdomen were also normal. 1,25-dihydroxyvitamin D level was 110 pg/mL (normal range 15–90 pg/mL).
On reviewing the history again, it was inferred that the father had intermittent bony pains and had undergone Ilizarov’s procedure for limb lengthening. On examination, he had bowing of both lower limbs and multiple scars from the limb lengthening procedure that he had undergone. This led us to think of a probable genetic etiology, and we worked her up further. The serum phosphate levels of both parents were low. The mother did not have any limb deformity. The calculated ratio of the renal tubular maximum reabsorption rate of phosphate to glomerular flitration rate (Tmp/GFR) was 1.8 mg/dL (normal range 3.2–5.1 mg/dL at 1–5 years). The tubular phosphate reabsorption was low at 75% (normal range 85–95%). The child was given oral phosphorus (15 mg/kg/day) and oral calcitriol (0.25 µg) once daily. A clinical exome sequencing done revealed a c.1965+1G>A (S’ splice site) heterozygous mutation in the PHEX gene on intron 19, a likely pathogenic mutation causing X-linked dominant hypophosphatemic rickets.
DISCUSSION
Hypophosphatemic rickets is an X-linked dominant disorder arising from a pathogenic variation in the PHEX gene and leads to an elevated concentration of circulating FGF23. The PHEX locus-specific database contains information on over 800 disease-causing variations in the PHEX gene found in XLH patients.[5] A cross-sectional study was done by Marik et al. in India at a single center, to identify the clinical characteristics and underlying genetics of patients with hypophosphatemic rickets. Twenty-two (33%) of the 66 patients enrolled with hypophosphatemic rickets had PHEX gene variations.[3] The mutation c.1965+1G>A, at intron 19, occurs at splicing acceptor/donor sites and has been reported from a study in South Korea and Spain, but we did not find a report of a similar mutation from the Indian subcontinent.[6,7] Marik et al. have reported a variation in the PHEX gene, c.1965 from North India; however, it was at exon 19.[3] Genetic variations hamper inactivation of FGF23 in the PHEX gene. Hypophosphatemia is caused by the loss of phosphorus at the proximal renal tubule as a result of elevated FGF23 levels.[2]
Nutritional rickets is by far the most common cause of rickets and is caused by vitamin D and/or Ca insufficiency. Non-nutritional rickets should be considered if there is a lack of response to adequate nutritional replacement for 2–3 months. The same can be suspected earlier in the presence of other clues such as alopecia, features of malabsorption, a strong family history, hypokalemia, metabolic acidosis, nephrocalcinosis, or dense bones on radiographs. Non-nutritional rickets results from conditions that either cause renal phosphate loss (hypophosphatemic rickets) or due to genetic disorders of vitamin D metabolism (VDDR).[2] Salient features differentiating nutritional, hypophosphatemic rickets and vitamin D-dependent rickets have been highlighted in Table 1.
| Laboratory test | Nutritional rickets | Hypophosphatemic rickets | Vitamin D-dependent rickets |
|---|---|---|---|
| Serum phosphorus | Low | Low | Low |
| Serum calcium | Low/Normal | Normal | Low |
| ALP levels | High | High | High |
| PTH levels | High | Normal | High |
| 25 hydroxyvitamin D | Low | Low/Normal | Variable |
| 1,25 dihydroxyvitamin D | High | Normal/High | Very high/Low |
ALP: Alkaline phosphatase, PTH: Parathyroid hormone
In XLH, craniotabes and rachitic rosary are uncommon, unlike in vitamin D deficiency. Frontal bossing may first appear in infants as young as six months. Clinical features such as waddling gait and bow legs become evident in early childhood when children with XLH begin to bear weight. Myalgia is not very apparent in these children. Lower limbs are more involved than upper limbs, so disproportionate short stature is almost always present unless treated early. XLH in childhood is portrayed by bone deformities such as genu valgum alone or in combination with other lower limb deformities, dental abscesses, bone pain, hearing difficulties, or craniosynostosis.[2] Identifying family members with features of XLH and early screening can lead to the detection of XLH even before the rachitic features are noticeable.
Children with XLH have normal serum Ca levels, elevated serum ALP activity, normal or raised PTH levels, hypophosphatemia with limited renal phosphate reabsorption, abnormally normal or lowered 1,25-dihydroxyvitamin D levels, and elevated FGF23 levels.[8] Radiographic features in children with XLH are different from those in nutritional rickets. The distinctive features are that the weight-bearing lower limbs, particularly the medial (genu varum) and lateral condyles (genu valgum), are more affected. Coarse trabeculations and thicker cortices are also prominent on radiographs.[2] Confirming the pathogenic variant causing rickets through genetic testing forms the gold standard in diagnosing the cause of hypophosphatemic rickets. Genetic testing not only aids in confirming the diagnosis but also helps finalize the management protocol, counsel regarding the prognosis, and plan further pregnancies.[2]
XLH is a multisystemic disorder requiring a multidisciplinary approach alongside subspecialties. These kids may experience severe side effects such as oral abscesses, hearing loss, increasing bone deformities, and craniosynostosis. Rather than bringing the serum phosphate concentration back to normal, the medical management aims to improve the osteomalacia, rachitic malformations, and maximize growth in the affected infants.
Phosphate supplementation remains the mainstay of the management. Most children benefit from taking a 20– 30 ng/kg/day calcitriol supplement since FGF23 inhibits the production of 1,25-dihydroxy cholecalciferol. Urinary Ca levels should be closely watched when taking calcitriol supplements to prevent nephrocalcinosis. When rickets heals, blood ALP levels return to normal, making it a trustworthy indicator of biochemical activity during follow-up.
Early diagnosis and treatment, which is begun before the child starts walking can prevent lower limb deformities and ensure better growth. Still, a significant drawback of traditional therapy is the observed uneven response to treatment, with many children failing to normalize their growth. Gastrointestinal symptoms, including loose stools and abdominal pain, as well as endocrine issues such as hypercalcemia, secondary hyperparathyroidism and renal problems, can often arise in children on long-term conventional treatment. FGF23 and phosphate supplements, which simulate the parathyroid cells over time, as well as the suppression of 1,25-dihydroxyvitamin D in those who are not receiving active calcitriol, can all result in secondary hyperparathyroidism, which subsequently increases phosphaturia and bone resorption. Conversely, insufficient phosphate consumption and immoderate consumption of active vitamin D may lead to lowered PTH levels and decreased bone turnover, which may affect the healing process of rickets. Hence PTH level should be monitored and kept within normal limits (10–65 pg/mL).[8] It has been demonstrated that burosumab, a recombinant human IgG1 monoclonal antibody that targets FGF23, considerably raises Tmp/GFR and serum phosphate levels. It has also been linked to improved physical function, decreased bone pain, and quick recovery of radiologic signs of rickets in patients with XLH. Studies have shown better linear growth in those patients treated with burosumab.[9,10]
Despite the initiation of medical management, children with XLH tend to develop progressive limb malalignments such as genu valgus or genu varus. Fatigue, bone pain, and lower limb and back muscle insufficiency are frequently linked to irregularities in gait. Even with very young children who have XLH, hemiepiphysiodesis is a safe and efficient procedure that can be used. At present, surgical treatments utilized to address angular knee abnormalities have unclear exact indications and primarily depend on the treating surgeon’s experience.[11]
Learning points
Not every case of rickets is caused by malnutrition, even in tropical nations.
When children exhibit severe rickets, pathological fractures, a positive family history, and an inability to respond to treatment with vitamin D supplementation, it is imperative to investigate the genetic causes of rickets seriously.
It is essential to diagnose and treat hypophosphatemic rickets early to prevent patient disability and decrease the burden on the family.
CONCLUSION
While the most common cause of rickets is a nutritional deficiency of Ca and vitamin D, there are also rarer “refractory” causes that affect the metabolism of phosphate and vitamin D. These must be taken into consideration based on clinical, biochemical, and radiographic findings so that the right treatment can be started as soon as possible. XLH is a rare, genetic, multi-systemic chronic illness that affects the quality of afflicted individuals’ lives. Manifestations of XLH could evolve and require the involvement of a multi-disciplinary team for effective management.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Genetic and clinical profile of patients with hypophosphatemic rickets. Eur J Med Genet. 2022;65:104540.
- [CrossRef] [PubMed] [Google Scholar]
- Mutational analysis of PHEX, FGF23 and DMP1 in a cohort of patients with hypophosphatemic rickets. Clin Endocrinol (Oxf). 2011;74:312-8.
- [CrossRef] [PubMed] [Google Scholar]
- Novel PHEX gene locus-specific database: Comprehensive characterization of vast number of variants associated with X-linked hypophosphatemia (XLH) Hum Mutat. 2022;43:143-57.
- [CrossRef] [PubMed] [Google Scholar]
- PHEX gene mutations and genotype-phenotype analysis of Korean patients with hypophosphatemic rickets. J Korean Med Sci. 2007;22:981-6.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic diagnosis of X-linked dominant hypophosphatemic rickets in a cohort study: Tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type. BMC Med Genet. 2011;12:116.
- [CrossRef] [PubMed] [Google Scholar]
- An overview of rickets in children. Kidney Int Rep. 2020;5:980-90.
- [CrossRef] [PubMed] [Google Scholar]
- X-Linked hypophosphatemic rickets: Multisystemic disorder in children requiring multidisciplinary management. Front Endocrinol (Lausanne). 2021;12:688309.
- [CrossRef] [PubMed] [Google Scholar]
- Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: A multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019;7:189-99.
- [CrossRef] [PubMed] [Google Scholar]
- Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3:R13-30.
- [CrossRef] [PubMed] [Google Scholar]




