
Translate this page into:
Allan-Herndon-Dudley syndrome: Report of a novel pathogenic variant in MCT8 gene

-
Received: ,
Accepted: ,
How to cite this article: Kulkarni A, Desai DR, Kothari PN. Allan-Herndon-Dudley syndrome: Report of a novel pathogenic variant in MCT8 gene. J Pediatr Endocrinol Diabetes 2022;2:78-80.
Abstract
X-linked MCT8 mutations cause Allan-Herndon-Dudley syndrome (AHDS) characterized by severe developmental delay and specific thyroid function abnormality. The report describes a 1 year-old boy with severe developmental delay, truncal hypotonia, quadriparesis (spastic), dystonia, and thyroid function abnormality (high free T3, low reverse T3, low free T4, and normal TSH) suggesting a form of impaired thyroid hormone sensitivity, namely, AHDS. WES analysis yielded novel hemizygous single-base pair duplication in exon 1 of MCT8 gene, validated by Sanger sequencing. MRI revealed generalized demyelination with poor gray and white matter differentiation and loss of brain volume. Indirect markers revealed thyrotoxicosis in some peripheral tissues. Having high index of suspicion for this rare disorder and plausible benefit of inclusion of free T3 assay in initial evaluation of infants and children with idiopathic developmental delay are emphasized. Amelioration of thyrotoxic features with tri-iodothyroacetic acid likely benefits patients at all ages. Neurodevelopmental improvement has been observed in those who received tri-iodoacetic acid early, which is being currently investigated.
Keywords
MCT8
Allan-Herndon-Dudley syndrome
SLC16A2 gene
Developmental delay
Tri-iodothyroacetic acid

INTRODUCTION
Deficiency of the thyroid hormone transporter, monocarboxylase tansporter 8 (MCT8) is associated with severe intellectual and motor disabilities as well as high serum T3 concentrations. Clinical diagnosis is often difficult and estimation of thyroid hormones especially free T3 (FT3) concentrations and appropriate genetic studies are necessary. Treatment using tri-iodothyroacetic acid formulations such as Triac that safely alleviates peripheral thyrotoxicosis, is recommended.
CASE REPORT
A One-year-old male presented with severe developmental delay, axial hypotonia, spastic quadriparesis, dystonia, and thyroid function abnormality (high free T3, low free T4, low reverse T3, and normal TSH) during evaluation. The initial generalized hypotonia, evolved into increased tone of the limbs, and development of dystonic movements were more pronounced over of the neck, limbs, and oromandibular region. Child was underweight (<3rd centile) and length measured between 10th and 25th centiles. Face appeared elongated with cupped ears. Resting heart rate varied between 100 and 120 beats/min (bpm), as confirmed on two occasions.
MRI brain depicted delayed myelination of cerebrum and cerebellum, impaired gray-white matter differentiation, and mild-to-moderate prominence of supra- and infratentorial sulci and ventricles, suggesting loss of brain volume [Figure 1]. Before referral, he had undergone karyotype, electromyography, urinary amino and organic acid profiles, plasma amino acid estimations, and auditory and visually evoked potentials, which were normal.
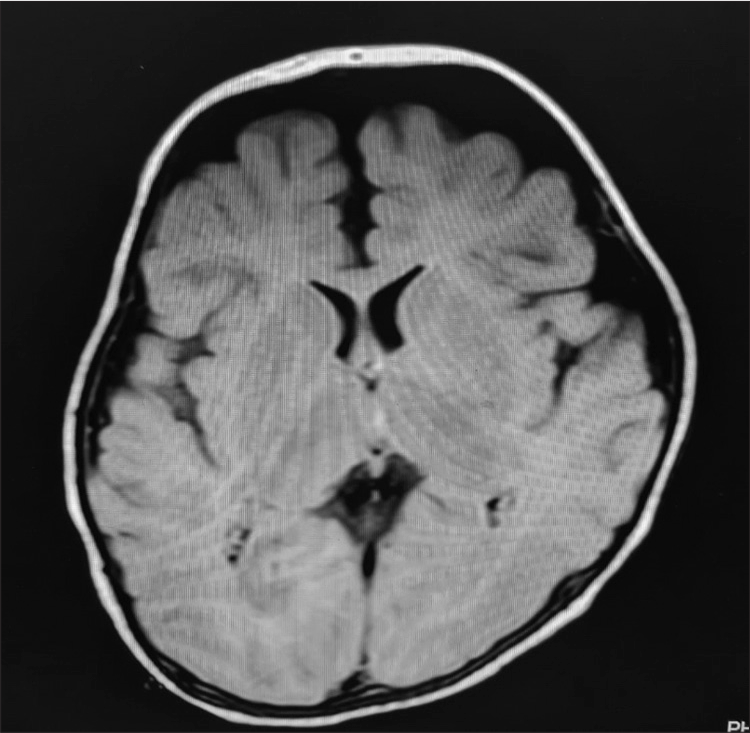
- MRI brain of the patient showing delayed demyelination of cerebrum and cerebellum, impaired gray-white matter differentiation and mild prominence of supra- and infratentorial sulci and ventricles.
A lower reverse T3 (rT3) level was documented on estimation. The clinical profile and abnormal thyroid functions implied impaired sensitivity to thyroid hormone, namely, Allan-Herndon-Dudley syndrome (AHDS). Discrepancy between length and weight centiles, myopenia, and mild but persistent tachycardia (HR ~100–120 bpm) suggested need for testing peripheral markers of thyroid function which suggested a state of increased catabolism in the liver (low cholesterol and high sex hormone binding globulin [SHBG])and at the muscle (high ammonia and lactate) as elaborated in [Table 1].
| Investigations | Laboratory results | Normal value ranges |
|---|---|---|
| TSH | 4.2 | 0.5–5.5 mIU/mL |
| Free T3 | 6.57 | 2.21–4.99 pg/mL |
| Free T4 | 0.71 | 0.8–2.0 ng/dL |
| Reverse T3 | 5.1 | 8.3–22.9 ng/dL |
| Lactate | 3.15 | 1–3.3 mmol/L |
| Ammonia | 46 | 1–40 umol/L |
| CPK | 152 | <120 μg/L |
| Total Cholesterol | 70 | 100–168 mg/dL |
| SHBG | 87 | 10–57 nmol/L |
Whole exome analysis by next-generation sequencing (NES) yielded a novel hemizygous single-base pair duplication in exon 1 of the MCT8 gene (c 407dupA) (p.Asn136LysfsTer31) which results in a frameshift and premature termination of protein 31 amino acids downstream of codon 136. This variant was not observed in either the 1000 Genomes or ExAC databases (Healthy population Database frequency <0.05). Bioinformatics algorithm MutationTaster2 predicted the variant to be deleterious. The boy’s mother was found to be a carrier of the same variant.
The patient has been initiated on treatment with oral tiratricol, that is, 3,3’,5-tri-iodothyroacetic acid (Triac) on an individual, off label basis at a starting dose of 175 μg/day (<10 kg body weight) and increments in daily dose by 175– 350 μg are being made every two-four weeks as per dose escalation protocol of Triac Trial I based on serum T3 concentrations (LCMS) to attain concentrations within a target range of 1.4–2.5 nmol/L initially.[1] The interval between T3 estimations for Triac dose titration is planned to be increased to 8–12 weekly after initial 12 weeks. Triac is currently not available commercially in India and is being provided on compassionate grounds to the patient by Rare Thyroid Therapeutics, Sweden.
Serum T3 concentrations <1.4 nmol/L may be permitted in children <2.5 years of age in the absence of dose-limiting toxicities, allowing higher Triac dosages, with a plausible added potential for neurocognitive improvement.[1]
The effects of Triac on clinical (heart rate by 24 h Holter) and biochemical features of thyrotoxicosis (serum T3 concentrations and tissue specific markers of thyroid hormone action) and neurodevelopment measured by Gross Motor Function Measure-88, Bayley’s Scale of Infant Development III, and MRI brain are planned after 48 weeks of Triac initiation, while body weight measurement, resting heart rate count (ECG), and neurologic examination are being done during each clinical visit to assess for improvements from baseline (Triac Trial II - MCT8-2019-2) (NCT02396459).
DISCUSSION
Deiodinases catalyzing production or degradation of thyroid hormone (TH) and tissue specific transporters determine biologic activity of TH. MCT8 is a specific thyroid hormone transporter and is widely expressed in brain, heart, kidney, adrenal glands, and thyroid. It is required for transporting thyroid hormone into neurons and thus neuronal development. MCT8 (SLC16A2) gene consists of six exons and is present on the X-chromosome (Xq13.2).
Although AHDS was first described in 1944, X-linked mutations in MCT8 gene responsible for the clinical phenotype were discovered only in 2004.[2] Presentation is initially characterized by generalized hypotonia, poor head-and-neck control followed by axial hypotonia, spastic quadriparesis, and dystonia. There is absence of speech development and communicative abilities are rudimentary. Elevated FT3, low rT3, low-normal or low FT4, and normal or slightly elevated TSH are biochemical hallmarks of AHDS.[3]
Large and smaller frameshift deletions, triplet or single amino acid deletions or insertions, and non-sense and missense mutations have been reported in the MCT8 gene till date. Both missense mutations and gene deletions cause equally severe psychomotor retardation.[2]
The key feature on neuroimaging in these patients appears to be delayed myelination, consistent with findings in our case. The presence of a plausible neurodegenerative process is suggested by the visible cortical atrophy.[2] A state of peripheral thyrotoxicosis in spite of neuronal T3 deficiency suggests possible presence of tissue specific TH transporters.[4]
Tri-iodothyroacetic acid is a thyroid hormone analog derived by conversion of T3 to its acetic acid derivative and its cellular entry is independent of MCT8. Tri-iodothyroacetic acid ameliorates thyrotoxic features in patients with MCT8 deficiency across ages and neurodevelopmental improvement is observed in the patients treated early.[1] Adverse effects of transient increase in perspiration and irritability were attributed to Triac during the Triac Trial I whereas serious adverse effects (intermittent infections) and hepatic insufficiency have been considered secondary to MCT8 deficiency and anticonvulsant drug, respectively, and unrelated to Triac.[1]
Effects of tri-iodothyroacetic acid on neurodevelopment and peripheral features of thyrotoxicosis in patients with MCT8 deficiency <2.5 years of age are being investigated by an international Phase IIb clinical trial (NCT02396459).[4]
CONCLUSION
The report makes a suggestion regarding having a high index of suspicion for the disorder and possible benefit of including FT3 assay in addition to TSH and FT4 while evaluating male infants and children with severe developmental delay of unknown etiology enabling timely diagnosis, plausible treatment, and requisite genetic counseling. The current TSH alone based neonatal screening tests fail to identify these patients though the extremely rare prevalence of this condition may not substantiate the case for including FT3 assay as a part of routine neonatal screening.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Effectiveness and safety of the triiodothyronine analogue Triac in children and adults with MCT8 deficiency: An international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019;7:695-706.
- [CrossRef] [Google Scholar]
- Severe neurological abnormalities in a young boy with impaired thyroid hormone sensitivity due to a novel mutation in the MCT8 gene. Hormones (Athens). 2017;16:194-9.
- [CrossRef] [Google Scholar]
- Monocarboxylate transporter 8 deficiency: Update on clinical characteristics and treatment. Endocrine. 2021;71:689-95.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term efficacy of T3 analogue Triac in children and adults with MCT8 deficiency: A real-life retrospective cohort study. J Clin Endocrinol Metab. 2022;107:e1136-47.
- [Google Scholar]




