
Translate this page into:
Pseudohypoparathyroidism type 1A and Albright hereditary osteodystrophy: A genetic conundrum of multiple hormonal resistances in a family


*Corresponding author: Anshika Mishra, Department of Pediatrics, Ganesh Shankar Vidyarthi Memorial Medical College, Kanpur, Uttar Pradesh, India. anshikamishra63796@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mishra A, Singh S, Mishra P. Pseudohypoparathyroidism type 1A and Albright hereditary osteodystrophy: A genetic conundrum of multiple hormonal resistances in a family. J Pediatr Endocrinol Diabetes. 2023;3:106-9. doi: 10.25259/JPED_19_2023
Abstract
We present a case series of pseudohypoparathyroidism type 1A (PHP1A) from a single family characterized by a notable pattern of inheritance and intrafamilial variability. The affected siblings exhibited clinical manifestations of PHP1A, including short stature, skeletal abnormalities, and hypocalcemia. Clinical exome sequencing revealed heterozygous inactivating mutations in the GNAS gene, leading to haploinsufficiency of the functional GNAS gene product. The proband, a 14-year-old male patient, demonstrated features such as short stature, obesity, skeletal anomalies, hypocalcemia, and hypothyroidism. The 12-year-old female sibling presented with bone pains, and carpopedal spasm with hypocalcemia without hypothyroidism. The mother had short 3rd and 4th metacarpals (Archibald sign), but biochemical profile was normal. This case report highlights the complex endocrine abnormalities and biochemical imbalances associated with PHP1A, emphasizing the genetic nature and variable expression of the disorder within the family.
Keywords
Pseudohypoparathyroidism 1A
Hypocalcemia
Hypothyroidism
GNAS gene
Archibald sign

INTRODUCTION
Pseudohypoparathyroidism type 1A (PHP1A) and Albright hereditary osteodystrophy (AHO) are rare genetic disorders characterized by multiple endocrine abnormalities and skeletal manifestations. PHP1A is a subtype of pseudohypoparathyroidism, which leads to impaired signaling of parathyroid hormone (PTH) in target tissues. AHO is a complex syndrome associated with PHP1A, characterized by round facies, short stature, and brachydactyly.[1]
One aspect of PHP1A and AHO is the occurrence of multiple hormonal resistances in affected individuals, which extends beyond PTH resistance. This resistance encompasses other hormones, such as thyroid-stimulating hormone (TSH), luteinizing hormone (LH), and growth hormone (GH). This unique hormonal resistance pattern can manifest in a familial context, affecting multiple siblings within the same family.[2]
In this paper, we delve into the genetic conundrum of PHP1A and AHO, focusing on the intricate relationship between these conditions, the underlying genetic mechanisms, and the perplexing phenomenon of multiple hormonal resistances in siblings.
CASE REPORT
Proband
The 14-year-old male patient presented with chief complaints of short stature, accompanied by a recent history of generalized seizures and documented hypocalcemia. The patient’s birth and developmental history were unremarkable. The patient had been treated with intravenous calcium during admission to a local hospital and was now referred for further evaluation. The patient had hypothyroidism diagnosed at the age of 3 years and was on levothyroxine replacement (50 µg daily). He is the second child of non-consanguineous Asian parents. An elder brother of the proband passed away during the neonatal period due to seizures, likely associated with hypocalcemia.
On physical examination, the boy weighed 58.5 kg (+0.86 standard deviation score [SDS]) and had a height of 138 cm (−2.63 SDS) and a body mass index (BMI) of 30.7 kg/m2 (+2.29 SDS; obese). The patient also exhibited a round face, short stubby fingers, Archibald sign (Short 3rd and 4th metacarpals), and brachymetacarpia of the 3rd and 4th toes on both feet [Figure 1]. The patient’s penile length was 10 cm, and bilateral testicular volumes were 16 mL. Tanner stage 4 of puberty, corresponding to 12.4–14 years, was also noted.

- Archibald sign in both siblings.
Table 1 displays the baseline endocrine and biochemical values obtained during the initial evaluation. GH stimulation test, which was also performed for the evaluation of short stature was normal. As shown in Figure 2a, computed tomography (CT) scan of brain revealed bilateral basal ganglia calcification. The electroencephalogram was normal. The findings highlight the complex endocrine abnormalities and biochemical imbalances observed in the patient, prompting further investigation into the underlying etiology of the presenting symptoms and laboratory findings.
| Proband | Sibling | Mother | Reference value | |
|---|---|---|---|---|
| Total calcium; mg/dL | 5.5 | 3.7 | 8.9 | 8.5–10.5 |
| Ionized calcium; mmoL/L | 0.72 | 0.38 | 1.18 | 1.14–1.29 |
| Inorganic phosphorus; mg/dL | 9.4 | 6.3 | 3.9 | 3.7–4.5 |
| Magnesium; mEq/L | 1.7 | 1.8 | 1.6–3.5 | |
| Alkaline phosphatase; U/L | 468 | 532 | 112 | 33.0–123.0 |
| Intact PTH; pg/mL | 221 | 103 | 15 | 10.0–65.0 |
| TSH; mU/mL | 13.2 | 0.8 | 0.6 | 0.3–5.0 |
| T4; mg/dL | 5.7 | 7.8 | 8.1 | 4.9–13.9 |
| LH; ng/mL | 20 | 17 | 25 | M: 15.0–60.0 |
| F: 10.0–40.0 | ||||
| FSH; mU/mL | 12.2 | 14.2 | 8.6 | 5.0–20.0 |
| Testosterone; ng/dL | 375 | - | - | M: 250.0–900.0 |
| Estradiol; pg/mL | - | 40.2 | - | F: 30.0–350.0 |
| Peak GH level after GH stimulation test; pg/mL | 13.5 | 12.8 | >10 |
PTH: Parathyroid hormone, TSH: Thyroid-stimulating hormone, FSH: Follicle-stimulating hormone, and GH: Growth hormone, LH- Luteinizing hormone, T4: Thyroxine
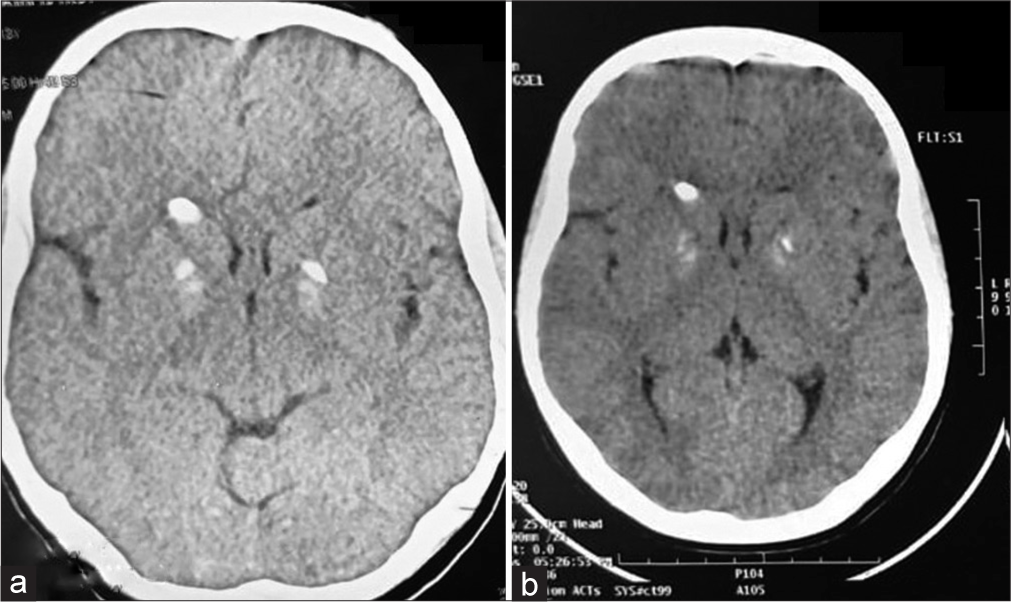
- (a) Basal ganglia calcification depicted in contrast-enhanced computed tomography (CECT) brain of the boy. (b) Basal ganglia calcification depicted in the CECT brain of the girl.
Sibling
A 12-year-old female sibling of the proband presented with bone pains and carpopedal spasms accompanied by positive Chvostek’s and Trousseau’s signs. Biochemical evaluation revealed that her symptoms were attributable to hypocalcemia. She received treatment with calcium and vitamin D supplements and was referred for further evaluation. Her birth and developmental history were unremarkable.
During physical examination, she had a height of 135 cm (−1.86 SDS) and weighed 35 kg (−0.60 SDS). She displayed characteristic features such as a round face, crowded dentition, bilateral broad thumbs, and brachymetacarpia of the 4th and 5th fingers [Figure 1]. Sexual maturity rating assessment indicated breast development (B2), absence of axillary hair (A), and prepubertal pubic hair (P1). X-ray imaging of her limbs revealed bilateral shortening of the 4th and 5th metacarpals and metatarsals. CT scan of brain depicted bilateral basal ganglia calcifications [Figure 2b]. Baseline endocrine and biochemical values of the patient showed notable differences compared to her brother. Unlike her brother, she did not exhibit hypothyroidism and had not required thyroxine supplementation.
Symptomatic hypocalcemia in both siblings was corrected by intravenous calcium gluconate and later oral calcitriol, along with calcium supplements. In a follow-up period of 1-year post-discharge, both patients remained symptom-free. The growth velocity of the proband was noted to be 6 cm/year and 5.8 cm/year for the sibling. The weight of proband decreased to 53.5 kg with a BMI of 25.7 kg/m2. Tanner’s staging of proband was stage 5, and that of the sibling was stage 3. Serum TSH was in the lower half of the normal reference range, and free T4 level was in the upper half of the normal reference range on thyroid supplementation in the proband. Serum calcium and phosphorus were in the normal reference range with urine calcium to creatinine ratio of 0.04–0.07 on 0.75 µg of calcitriol, 750 mg of oral calcium carbonate, and 600 IU of cholecalciferol. The follow-up plan is periodic monitoring with titration of medications to maintain serum calcium levels in the lower normal range with a normal urine spot calcium/creatinine ratio.
The mother exhibited characteristic shortening of the 3rd and 4th metacarpal and metatarsal bones. She was also evaluated, including the biochemical and hormonal profiles. The results showed normal thyroid function test results, as well as normal levels of follicle-stimulating hormone, LH, PTH, calcium, and phosphate. Unfortunately, genetic analysis of the parents could not be performed due to financial constraints.
The siblings demonstrated clinical manifestations of the disorder, as shown in the third-generation pedigree chart [Figure 3]. The proband exhibited features consistent with PHP1A, including obesity, short stature, skeletal abnormalities, and hypothyroidism. The elder brother, who died in the neonatal age with multiple seizures, was likely having hypocalcemia, a common complication of PHP1A. This familial pattern underscores the genetic nature of PHP1A, with evidence of both vertical transmission from the mother to the affected offspring and the potential for intrafamilial variability in the expression of the disorder. The presence of skeletal abnormalities, hypocalcemic seizures, and familial recurrence strongly suggests a diagnosis of PHP1A in this family. To unravel the genetic basis underlying the PHP1A observed in the affected siblings, clinical exome sequencing by next-generation sequencing was performed, which yielded heterozygous mutation in the GNAS gene (NM_000516.4[GNAS]: c2787_2788del[p.Val930AspfsTer12]) located on chromosome 20q13.2-13.3, in both the siblings.
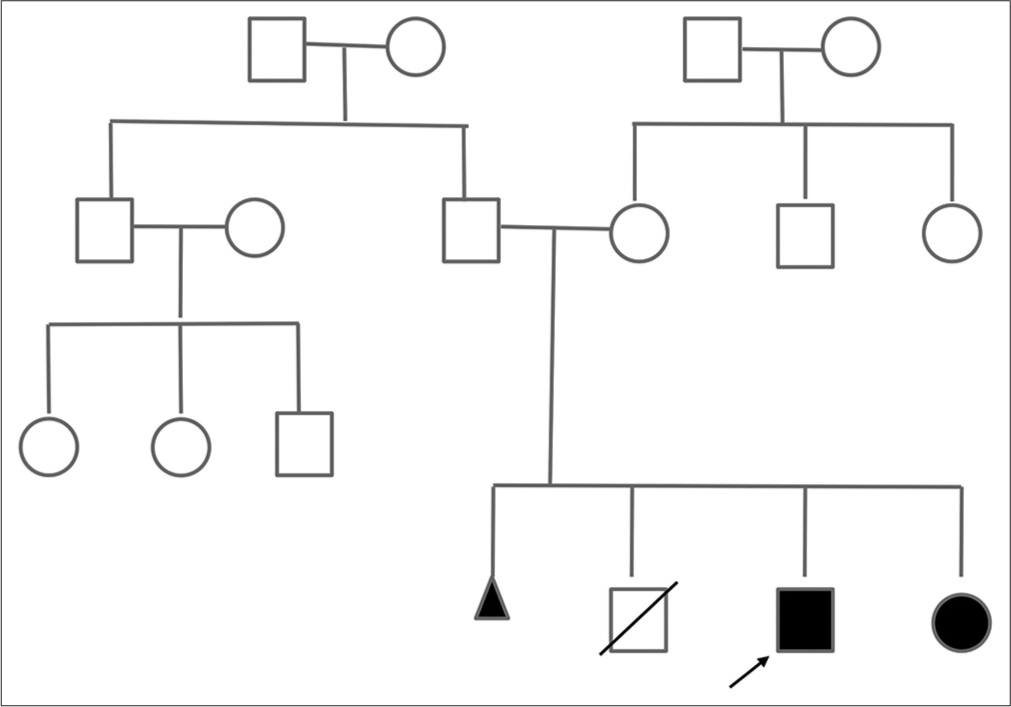
- Three generation pedigree. Clear box: Unaffected male, Clear circle: Unaffected female, Black box: Affected male, Black circle: Affected female, Diagonal line through box: Deceased male, Black triangle: Stillbirth.
7DISCUSSION
PHP is a rare disorder characterized by a set of clinical and biochemical abnormalities, including hypocalcemia, hyperphosphatemia, and elevated PTH levels. PHP can be categorized into different subtypes based on various factors such as inheritance pattern, genetic mutation, sites of PTH resistance, and the presence of AHO features.[3] Among these subtypes, PHP1A is distinguished by the presence of AHO characteristics and impaired responsiveness of target tissues to hormones that signal through Gs-coupled receptors, including PTH, TSH, gonadotropins, and GH-releasing hormone.
Individuals with PHP1A exhibit features of AHO, such as short stature, round face, and skeletal abnormalities, along with characteristic hormonal resistance. The underlying genetic abnormality responsible for PHP1A involves heterozygous inactivating mutations in the GNAS gene. These mutations lead to a deficiency in the GNAS protein, resulting in disrupted signaling pathways and reduced responsiveness of target tissues to hormonal stimulation.[4]
The clinical presentation of PHP1a can vary widely among affected individuals, even within the same family.[5] In the presented case series, we observed different clinical manifestations among the siblings, including short stature, skeletal abnormalities, and hypocalcemia. The deceased neonatal sibling likely succumbed to seizures associated with severe hypocalcemia, highlighting the potential complications of this disorder.
Management of PHP1A focuses on addressing the underlying hormonal imbalances and associated symptoms. This typically involves calcium and vitamin D supplementation to correct hypocalcemia and normalize serum calcium levels. In some cases, additional hormone replacement therapies may be required to manage specific endocrine deficiencies, such as thyroxine replacement for hypothyroidism.[6]
The intrafamilial variability observed in our case series raises intriguing questions regarding the potential influence of modifier genes and epigenetic factors on disease expression. The unaffected father and the presence of non-penetrant or mild phenotypes in the parents suggest the involvement of modifying factors that may modulate the severity of PHP1A within the family.[7]
Genetic counseling is crucial for families affected by PHP1A to provide accurate recurrence risk assessment and guide family planning decisions. It is important to educate patients and their families about the need for regular monitoring, individualized treatment strategies, and potential long-term complications associated with PHP1A.
CONCLUSION
Our familial case series sheds light on the complex nature of PHP1A, encompassing genetic, clinical, and intrafamilial variability. Understanding the underlying genetic abnormalities, recognizing the clinical manifestations, and implementing appropriate management strategies are essential for optimizing patient care and genetic counseling in individuals and families affected by PHP1A. Further research is needed to elucidate the role of modifier genes and epigenetic factors in the pathogenesis and variable expression of this disorder.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
Patient’s consent is not required as patient’s identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Pseudohypoparathyroidism In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2023.
- [Google Scholar]
- Nonclassic features of pseudohypoparathyroidism type 1A. Curr Opin Endocrinol Diabetes Obes. 2017;24:33-8.
- [CrossRef] [PubMed] [Google Scholar]
- Pseudohypoparathyroidism In: Scriver CR, Beardet AL, Valle D, Sly WS, eds. The metabolic and molecular bases of inherited diseases. Vol 8. New York: McGraw-Hill; 1965. p. :4205-17.
- [Google Scholar]
- Resistance to multiple hormones in patients with pseudohypoparathyroidism. Association with deficient activity of guanine nucleotide regulatory protein. Am J Med. 1983;74:545-56.
- [CrossRef] [PubMed] [Google Scholar]
- A patient with pseudohypoparathyroidism type 1a previously misdiagnosed as hereditary multiple exostosis: A case report. Exp Ther Med. 2022;24:597.
- [CrossRef] [PubMed] [Google Scholar]
- Management of pseudohypoparathyroidism. Curr Opin Pediatr. 2019;31:537-49.
- [CrossRef] [PubMed] [Google Scholar]
- Wind of change in pseudohypoparathyroidism and related disorders: New classification and first international management consensus. Endocrinol Diabetes Nutr (Engl Ed). 2018;65:425-7.
- [CrossRef] [PubMed] [Google Scholar]




