
Translate this page into:
Neurodevelopmental delay as the initial presentation of thyroid hormone resistance syndrome
 , Maria Del Carmen De Mingo Alemany2, Francisco Javier Maravall Royo1, Francisca Moreno Macián2, Pablo Abellán Galiana1, Agustín Ángel Merchante Alfaro1
, Maria Del Carmen De Mingo Alemany2, Francisco Javier Maravall Royo1, Francisca Moreno Macián2, Pablo Abellán Galiana1, Agustín Ángel Merchante Alfaro1

*Corresponding author: Esther Serisuelo Meneu, Department of Endocrinology and Nutrition, Hospital General Universitario Castellón, Castellón de la Plana, Spain. eserimeneu@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Serisuelo Meneu E, De Mingo Alemany MJ, Maravall Royo FJ, Moreno Macián F, Abellán Galiana P, Merchante Alfaro AA. Neurodevelopmental delay as the initial presentation of thyroid hormone resistance syndrome. J Pediatr Endocrinol Diabetes. 2024;4:93-7. doi: 10.25259/JPED_3_2024
Abstract
Thyroid hormone (TH) resistance syndrome is a genetic disorder usually caused by a defect in TH receptors. Moreover, transport and intracellular metabolism alterations have also been described. This case report presents a child with Allan–Herndon–Dudley syndrome characterized by a mutation on the X-linked monocarboxylate transporter 8 gene, a condition that affects the transport of THs across the cell membrane, leading to hypothyroidism in the central nervous system and hyperthyroidism in peripheral tissues, causing severe neurodevelopmental delay manifesting as generalized hypotonia from birth. The utility of monitoring thyroid functions, genetic testing, and triiodothyroacetic acid in the management is highlighted.
Keywords
Thyroid hormone resistance syndrome
Monocarboxylate transporter 8 gene
Allan–Herndon–Dudley syndrome
Neurodevelopmental delay
Triiodothyroacetic acid

INTRODUCTION
Thyroid hormone resistance syndrome (THRS) is a rare genetic disorder that is characterized by an alteration in sensitivity to thyroid hormones (THs) in peripheral tissues. This is generally caused by defects in nuclear TH receptors THRα and THRβ, which are located in different tissues of the body. Consequently, the phenotype of this syndrome can be very varied.[1] Other uncommon hereditary disorders affect the secretion, transport, and metabolism of TH.
The only known alteration in the transport of TH across the cell membrane, named Allan– Herndon–Dudley syndrome (AHDS), is caused by a mutation in the monocarboxylate transporter 8 (MCT8 gene), a specific transporter for TH that is linked to the X-chromosome with 100% penetrance in males and an estimated prevalence of 1:70,000.[2] This syndrome alters the function of the MCT8 transporter, reducing the availability of TH in the brain and, therefore, leading to hypothyroidism in the central nervous system. This deficit is not compensated by other TH transporters. As a result, a severe psychomotor deficit with manifest hypotonia from the 1st days of life occurs. This syndrome is characterized by low levels of thyroxine (T4) with normal or increased thyroid-stimulating hormone (TSH), elevated triiodothyronine (T3), and decreased levels of reverse T3 (rT3).[3]
CASE REPORT
A 3-month-old male child was born healthy at 38 weeks of gestation with a normal vaginal delivery. He presented a normal APGAR score, normal birth anthropometry, and a negative screening test for congenital hypothyroidism with no known family history of thyroid disorders.
He was hospitalized for further evaluation of his generalized hypotonia during the 1st month of life. The TH profile showed normal TSH 1.76 mU/L (0.350–4.940 mU/L), low T4 0.58 ng/dL (0.7–1.48 ng/dL), and a negative test for thyroid autoimmunity. He was initially diagnosed as a child with central hypothyroidism. The rest of the pituitary hormones were within the normal range. The MRI of the brain did not show any apparent structural abnormalities of the hypothalamic-pituitary axis, but there was a delay in the brain myelination process. A thyroid ultrasound revealed a small thyroid gland without any focal abnormalities.
The child was initially started on treatment with levothyroxine at a low dose, which was slowly escalated under clinical and biochemical monitoring to 150 µg/day (18.75 µg/kg/day) at 1 year of age. With this, free T4 was normalized to 0.88 ng/dL (0.7–1.48 ng/dL) with a persistently suppressed TSH <0.00 mU/L (0.350–4.940 mU/L) [Figure 1].
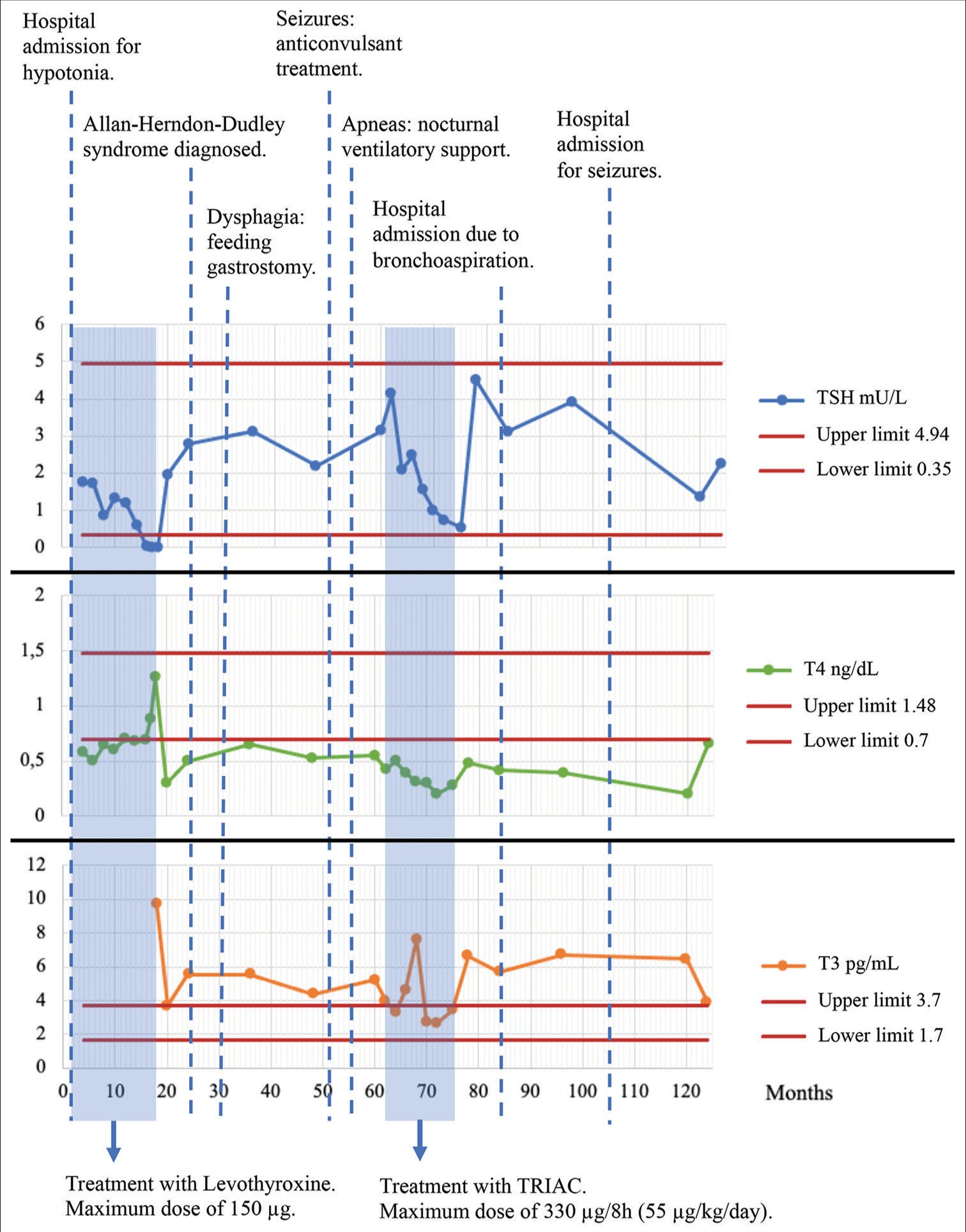
- Thyroid hormones and clinical evolution with the years. TRIAC: Triiodothyroacetic acid, TSH: Thyroid stimulating hormone, T4: Free thyroxine, T3: Free triiodothyronine.
On the follow-up, the patient progressed to severe psychomotor deficit with central hypotonia, especially in the neck, and inability to sit or walk with spastic quadriplegia, dystonia, and dyskinesia. He also showed speech alteration and severe dysphagia, requiring feeding gastrostomy when he was 2 years old. His condition worsened as he started getting episodes of nocturnal apneas and seizures, requiring follow-up by neurology colleagues to initiate anticonvulsant treatment.
In view of the poor response to treatment and worsening clinical profile, the thyroid axis was reevaluated, and T4 therapy was discontinued. A genetic test using massive DNA sequencing techniques and including 538 genes related to neurodevelopmental disorders was performed. The results revealed a mutation in the MCT8 gene (SCL16A2, p.Pro215Leu change on the X-chromosome), suggesting the diagnosis of AHDS. This mutation had not been previously described. However, prediction algorithms indicated a pathogenic effect. Moreover, the clinical manifestations presented by the patient were highly consistent with those described in AHDS, including the neurological condition and the alteration of the initial thyroid profile, as well as the poor response to treatment with levothyroxine.
After the diagnosis, TH levels were re-evaluated: elevated T3 4.76 pg/mL (1.70–3.70 pg/mL), low rT3 40 pg/mL (90–330 pg/mL), low T4 0.5 ng/dL (0.7–1.48 ng/dL), and elevated TSH 6.377 mU/L (0.350–4.940 mU/L). The high levels of sex-hormone-binding-globulin 152 nmol/L (16–86 nmol/L) and the decrease in total cholesterol 120 mg/dL (<200 mg/dL) were attributed to the effect of high concentrations of T3 in peripheral tissues. The TRH stimulation test results were normal, with adequate pituitary TSH stimulation [Figure 1].
The patient was proposed to start treatment with 3,3’,5-triiodothyroacetic acid (TRIAC), a TH analog that penetrates cell membranes without requiring the MCT8 transporter. TRIAC was started at the age of 5 years of age with increasing doses up to 330 µg every 8 h orally (52.8 µg/kg/24 h). The tolerance of the treatment was good and resulted in a reduction of spasticity and night apneas. However, treatment was suspended after 1 year in the absence of neurological improvement. TSH persisted in range at 0.6 mU/L (0.350–4.940 mU/L), T4 was low <0.4 ng/dL (0.7–1.48 ng/dL), and T3 presented elevated levels 4.73 pg/mL (1.70–1.48 ng/dL) [Figure 1].
The patient is now 13 years old, and he is being followed up by specialists in the digestive and nutrition unit (gastrostomy control and malnutrition), pulmonology (sleep apnea), neurology (seizures and nocturnal agitation), and endocrinology (thyroid disorder). He is currently under symptomatic treatment [Figure 2].
- Clinical photograph of the patient.
DISCUSSION
THRS includes many genetic defects with multiple phenotypic expressions, both clinical and biochemical. In the case of AHDS, the mutation in the X-linked MCT8 gene that expresses the TH transporter across the cell membrane (especially at the level of the central nervous system) explains the most characteristic symptoms appearing from the 1st month of life.[3]
MCT8 is a specific transporter of iodothyronines. To date, no other substrate for this transporter has been found. The transport rate of T3 and T4 (with preference of T3) is very high compared to other TH transporters, such as those of the NTCP and OATP families. MCT8 is expressed in the brain, heart, lungs, kidneys, thyroid, placenta, skeletal muscle, pituitary, and liver. In the brain, it can be detected, specially in neuronal pattern, choroid plexus, and blood-brain barrier.[4]
It should be noted that only the THRβ nuclear receptor is important in the regulation of the negative feedback system of the hypothalamic-pituitary-thyroid axis, maintaining TH concentration within the normal range. Therefore, the analytical pattern of AHDS differs from the pattern of THRS (THRβ mutation) with normal or elevated TSH and elevated T4 and T3. Hence, the diagnosis of AHDS may be deferred.[5]
In this case, the elevation in serum T3 is due to several reasons. Mutations in MCT8 may induce higher secretion of T3 and a lower of T4 for reasons not yet well known.[3] The elevated T3 levels increase deiodinase-1 in the liver and kidneys, which allows the conversion of T4 into T3 to further increase circulating T3 levels. Furthermore, the decrease in the entry of T3 into the brain and other tissues that express deiodinase-3 causes less degradation, which contributes to an increase in blood levels. In addition, a greater excretion of T4 by the kidney provokes a sustained decrease in T4. Finally, the low levels of rT3 are caused by an increment in its degradation by deiodinase-1 in the liver and kidneys and a lower production of rT3 in tissues such as the brain.[4]
The most common approaches to treat this condition are limited to symptomatic measures such as feeding gastrostomy to avoid severe weight loss and aspiration; use of orthopedic support; carbamazepine, levodopa, or anticholinergics to treat dystonia; anticonvulsants to treat seizures; and scopolamine or glycopyrrolate for drooling.
Treatment with levothyroxine is ineffective because it can increase T3 and peripheral thyrotoxicosis.[2] However, treatment with propylthiouracil (which inhibits deiodinase-1 and reduces T4 to T3 conversion) with levothyroxine at high doses has been described in the literature. It has been shown to be effective in improving thyroid biochemical profile and nutritional status.[6]
Other studies focus on TH analogs that use alternative transporters to substitute this genetic deficit.[7] 3,5-diiodoothyropropionic acid was tested in a pilot study with four children, improving biochemical abnormalities.[8] TRIAC also showed improvement in the clinical manifestations of thyrotoxicosis.[9] The main biochemical effect was the reduction of T3 with TSH suppression and, therefore, a decrease in the synthesis and secretion of TH. This improves hypermetabolism and its consequences. However, only slight improvement in terms of intellectual disability was observed in children under 3 years. In older children, there was no improvement.[2,7,9] Treatment with intrathecal TRIAC was also tested in mice. Although brain T3 availability increased, the results were not satisfactory.[10]
At present, an ongoing trial (NCT02396459) is investigating the effect of TRIAC treatment in children below 30 months of age. Patients are treated for 96 weeks, and the effect of treatment on neurodevelopmental impairment and peripheral thyrotoxicosis is evaluated.[9]
CONCLUSION
A thorough understanding of AHDS turned out to be essential to properly diagnose this boy with a delay in neurodevelopment and TH alteration. Genetic testing coupled with a thyrotropin releasing hormone (TRH) stimulation test and serial biochemical thyroid function helped in the proper diagnosis of the index case. An early diagnostic approach, ideally before birth and an appropriate treatment with new molecules such as TRIAC are the most important aspects to improve the neurological problems caused by this condition. The development of new, more effective, and efficient molecules with a better capacity to penetrate through the blood-brain barrier is still necessary for a better neurodevelopmental outcome.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. Thyroid. 2014;24:407-9.
- [CrossRef] [PubMed] [Google Scholar]
- Disease characteristics of MCT8 deficiency: An international, retrospective, multicentre cohort study. Lancet Diabetes Endocrinol. 2020;8:594-605.
- [CrossRef] [PubMed] [Google Scholar]
- Mice deficient in MCT8 reveal a mechanism regulating thyroid hormone secretion. J Clin Invest. 2010;120:3377-88.
- [CrossRef] [PubMed] [Google Scholar]
- Transporte y acción de las hormonas tiroideas en cerebro de ratón: Interrelación entre Mct8 y Lat2 Madrid: Universidad Autónoma de Madrid; 2013.
- [Google Scholar]
- Do clinical manifestations of resistance to thyroid hormone correlate with the functional alteration of the corresponding mutant thyroid hormone-beta receptors? J Clin Endocrinol Metab. 1995;80:3246-56.
- [CrossRef] [PubMed] [Google Scholar]
- Beneficial effects of propylthiouracil plus L-thyroxine treatment in a patient with a mutation in MCT8. J Clin Endocrinol Metab. 2008;93:2084-8.
- [CrossRef] [PubMed] [Google Scholar]
- MCT8 deficiency: The road to therapies for a rare disease. Front Neurosci. 2020;14:380.
- [CrossRef] [PubMed] [Google Scholar]
- Diiodothyropropionic acid (DITPA) in the treatment of MCT8 deficiency. J Clin Endocrinol Metab. 2012;97:4515-23.
- [CrossRef] [PubMed] [Google Scholar]
- Effectiveness and safety of the tri-iodothyronine analogue Triac in children and adults with MCT8 deficiency: An international, single-arm, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019;7:695-706.
- [CrossRef] [PubMed] [Google Scholar]
- Intracerebroventricular administration of the thyroid hormone analog TRIAC increases its brain content in the absence of MCT8. PLoS One. 2019;14:e0226017.
- [CrossRef] [PubMed] [Google Scholar]




