
Translate this page into:
Neonatal hypoglycemia: A review with focus on practical challenges and recent updates on management


*Corresponding author: Santhosh Olety Sathyanarayana, Department of Paediatric and Adolescent Endocrinology, Karnataka Institute of Endocrinology and Research, Bengaluru, Karnataka, India. docsanthosh@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sathyanarayana SO, Parikh AC, Sowjanya GT. Neonatal hypoglycemia: A narrative review with a focus on practical challenges and recent updates on management. J Pediatr Endocrinol Diabetes. 2024;4:84-92. doi: 10.25259/JPED_20_2024
Abstract
At birth, a neonate undergoes a transition from the continuous maternal supply of glucose to a variable and intermittent oral glucose intake, which is regulated by the interplay of hormones and metabolic enzyme induction. Because low plasma glucose concentrations are common in the neonatal period, it may be difficult to identify those who have pathologic hypoglycemia. Hence, it is important to formally evaluate such babies by drawing critical samples. Here, we present two cases of neonatal hypoglycemia where the presentation had some similarities, but the comprehensive evaluation revealed a varied etiological spectrum necessitating lifelong management. Through these case studies, authors discuss practical challenges in the diagnosis, management, and follow-up of neonates with endocrine causes of hypoglycemia.
Keywords
Congenital hyperinsulinism of infancy
Hypoglycemia
Neonatal
Hyperinsulinemia
Hypopituitarism

INTRODUCTION
Hypoglycemia is a common metabolic emergency often related to the physiological transition of babies postnatally but rarely attributable to pathological causes, which are often missed. Glucose homeostasis in neonates is mainly dependent on the interplay of insulin, cortisol, and growth hormone (GH). Cortisol deficiency may present with severe hypoglycemia, cholestasis, failure to thrive, hyponatremia without hyperkalemia, fluid imbalance, temperature instability, a lack of thymic involution, and rarely heart failure.[1,2] The most common endocrine causes include congenital hyperinsulinemia, stress-induced hyperinsulinemia, and hypopituitarism.[3] Critical sampling helps in the identification of endocrine disorders, which warrant further confirmation. We describe two cases of severe neonatal hypoglycemia with different etiologies, aiming to review recent literature and discuss practical challenges that arise during diagnosis, management, and monitoring of these infants.
CASE SERIES
Case 1
A 2-day-old baby girl, born of a non-consanguineous union and with a birth weight of 3 kg, presented with hypoglycemic seizures (capillary blood glucose [BG] 26 mg/dL), and treated symptomatically with intravenous (IV) dextrose. In view of the persistent and recurrent hypoglycemic convulsions, the baby was evaluated in a pediatric endocrine unit, and critical samples revealed BG 28 mg/dL, plasma insulin 18.9 µU/L, serum ketones 0.3 mmol/L, serum cortisol 18.5 µg/dL (normal 2–11 µg/dL), plasma ammonia 54 µmol/L (9–30 µmol/L), and a positive glycemic response to subcutaneous (SC) glucagon injection. The neonate was managed with oral diazoxide (20 mg/kg/d) and octreotide SC injections (11 µg/kg/d), resulting in normalization of BG by 3 weeks. Next-generation sequencing (NGS) revealed a paternally inherited dominant ABCC8 mutation at exon 36. An 18F-DOPA positron emission tomography/computed tomography (PET/CT) nuclear scan showed a diffusely increased uptake in the pancreas [Figure 1], which was confirmed by a Gallium-68 DotaExendin-4 scan [Figure 1]. There was a history of early-onset diabetes mellitus in the father (age of onset 31 years) and paternal grandfather, managed with oral hypoglycemic agents. At the last follow-up, the child was 3 years old, and her growth (weight and height on the 10th percentile on the World Health Organization charts) and development were in normal ranges. The infant remains euglycemic with occasional hypoglycemia on SC octreotide at 8 µg/kg/d in three divided doses.
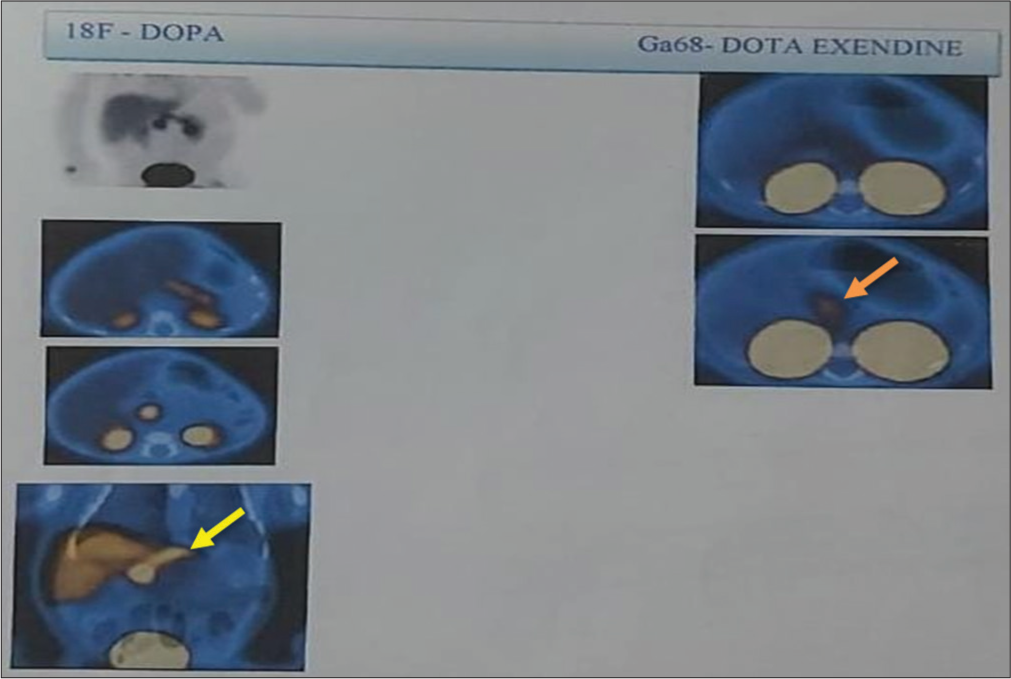
- 18F-DOPA positron emission tomography – Diffuse uptake in the pancreas with SUV 20.3 (Yellow Arrow). No obvious focal lesion in the pancreas. Gallium-68 Dota Extendin-4 scan – Diffuse uptake seen in the pancreas with SUV 3.7 (Orange Arrow). 18F-DOPA: 18 Fluoro-dihydroxyphenylalanine, SUV: Standardized uptake value.
Case 2
A baby girl born of a non-consanguineous marriage with a birth weight of 2.9 kg and a length of 48 cm by lower-segment cesarean section with a breech presentation developed symptomatic hypoglycemia on day 1 of life and was treated with IV dextrose. The baby did not have further hypoglycemic episodes and received phototherapy for unconjugated hyperbilirubinemia. Cord thyroid-stimulating hormone (TSH) was 0.03 mIU/L; hence, repeated on day 8, it was TSH 0.04 mIU/L with free thyroxine 0.2 ng/dL. The baby was readmitted in the 2nd week of life with hypoglycemia (BG 42 mg/dL) and unconjugated hyperbilirubinemia (17 mg/dL). The critical sample showed cortisol 10 µg/dL, adrenocorticotropic hormone (ACTH) 15 pg/mL, ketones and insulin suppressed, and GH 0.03 ng/mL. She was started on hydrocortisone, and in view of persistent hypoglycemia, the dose was titrated to a maximum of 20 mg/m2/d in three divided doses, followed by the addition of levothyroxine (LT4), 37.5 µg/d. In spite of optimal replacement of hydrocortisone and LT4, the baby continued to have hypoglycemic episodes for the next 3 days, and GH therapy was started at 0.7 mg/m2/d, and euglycemia was achieved within 3–4 days after initiation. MRI of the pituitary revealed a cystic lesion of 6.5 × 7.9 × 9.2 mm size within the sella and suprasellar region, significantly compressing the pituitary gland, suggestive of Rathke’s cleft cyst with normal posterior pituitary (PP) bright spot [Figure 2]. The ophthalmology evaluation was normal. Neurosurgery advice was conservative management with a repeat imaging after 1 year. NGS revealed a compound heterozygous mutation at exons 4 and 2 of the LHX3 gene in the index patient. Parental testing revealed an asymptomatic heterozygous missense variation in exon 4 of the LHX3 gene in the father and heterozygous missense variation in exon 2 of the LHX3 gene in the mother by Sanger sequencing. At 6 months of life, insulin-like growth factor binding protein-3 was 1.53 µg/mL (normal 0.7–3.6). Follow-up MRI brain at 2 years of age showed resolution of previously seen Rathke’s cyst with hypoplastic pituitary (pituitary height 2.8 mm and thin stalk of 1.2 mm). Features of Chiari 1 malformation with peg-like cerebellar tonsils herniating into the posterior cervical spinal canal and indenting the cord were seen without any evidence of edema. At 2 year follow-up, the baby was doing well on GH 0.85 mg/m2/d, LT4 37.5 µg/d, and hydrocortisone 2.8 mg/m2/d. It was planned to wean off hydrocortisone with subsequent adrenal axis evaluation, but unfortunately, the baby succumbed to an intercurrent lung infection. Parents have opted for prenatal genetic testing following counseling.
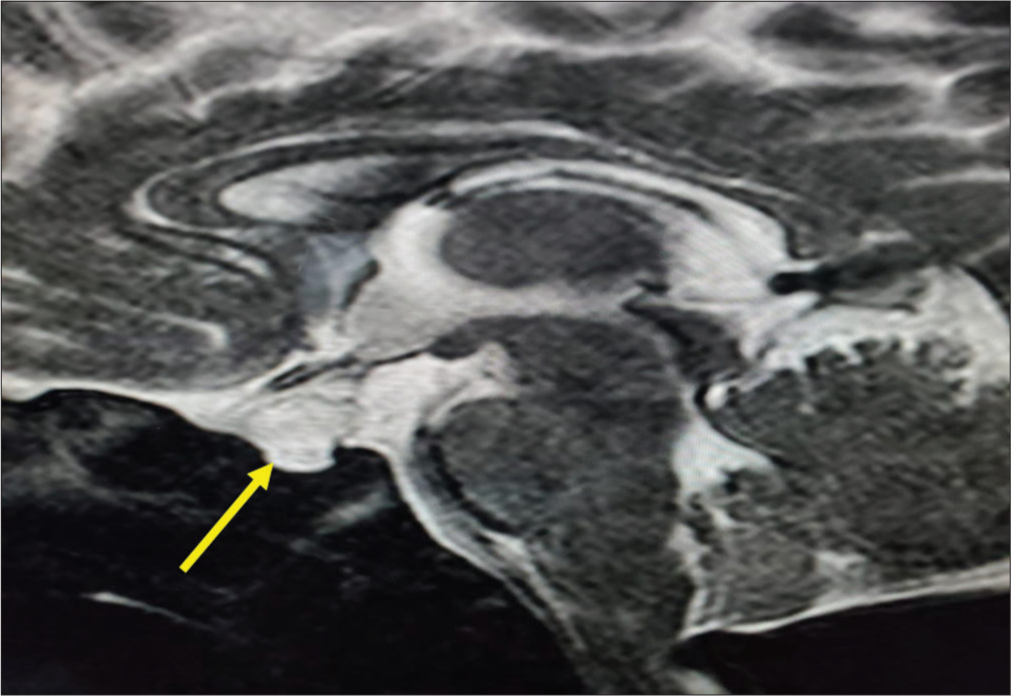
- Cystic lesion within the sellar and suprasellar region significantly compressing the pituitary gland (Yellow Arrow).
DISCUSSION
The approach to newborns with hypoglycemia persisting beyond the initial days is depicted in Figure 3. Congenital hyperinsulinism of infancy (CHI) is one of the most common causes of persistent and recurrent hypoglycemia in the newborn. In a resource-limited setting, difficulties pertaining to evaluation are compounded by the high cost and non-availability of medications, unresponsiveness to medical therapy, limited facilities for nuclear imaging, and only a few tertiary centers having surgical expertise in managing the condition.
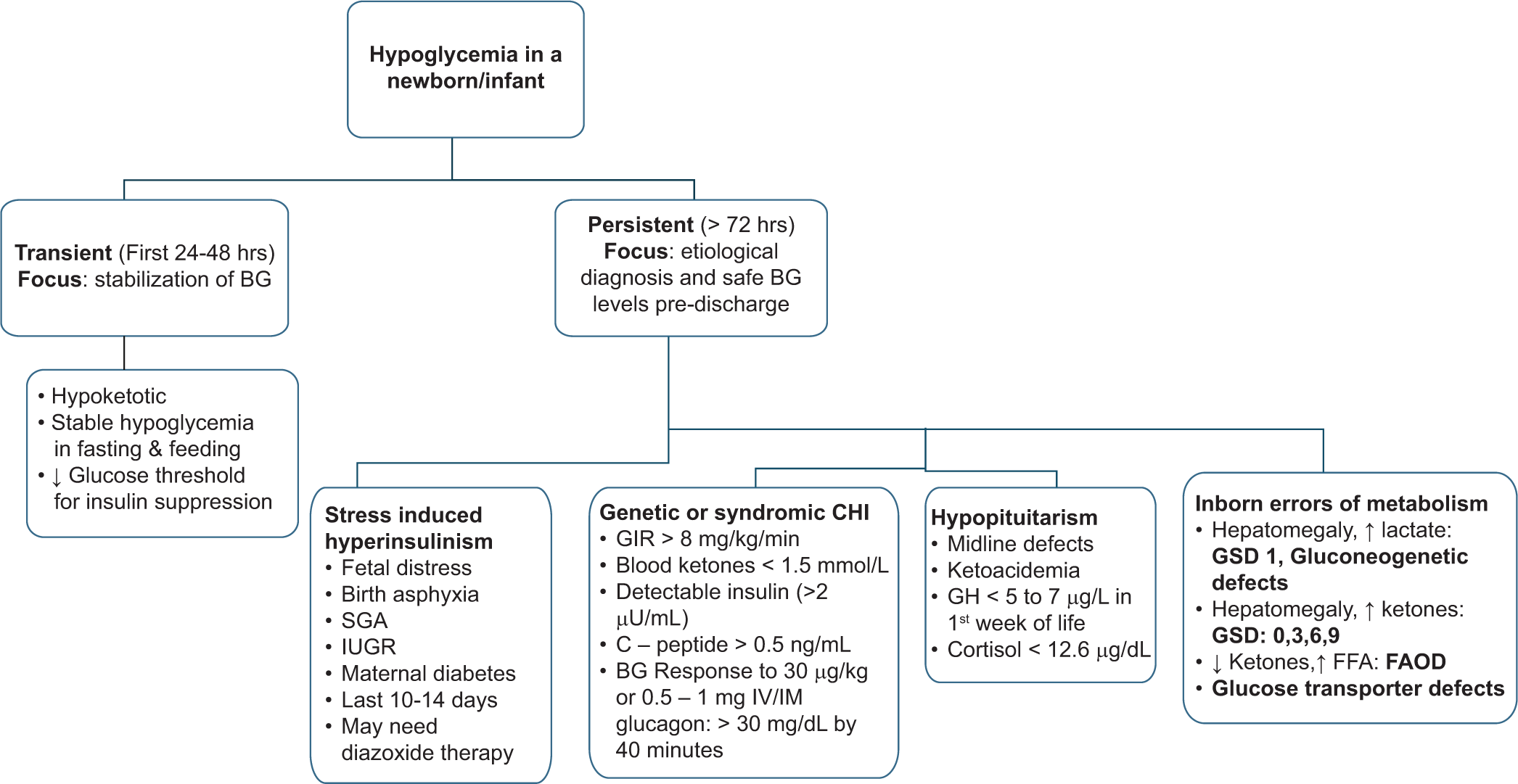
- Approach to a newborn/infant with hypoglycemia. BG: Blood glucose, SGA: Small for gestational age, IUGR: Intrauterine growth restriction, CHI: Congenital hyperinsulinism, GIR: Glucose infusion rate; IV: Intravenous, IM: Intramuscular, SC: Subcutaneous, GH: Growth hormone, GSD: Glycogen storage disease, FFA: Free fatty acids, FAOD: Fatty acid oxidation defect.
The approach to CHI in the developed world begins with a rapid analysis of the most frequently implicated (60%) CHI genes, namely, ABCC8 and KCNJ11.[4,5] These genes encode the two subunits of the ATP-sensitive potassium channel (K-ATP), namely, SUR-1 and Kir6.2, respectively, and are located next to each other on chromosome 11p15. The various patterns of inheritance of their mutations and their clinical implications are shown in Table 1. Rapid gene sequencing, with a turnover time of <10 days, confirms the diagnosis along with identification of the possibility of focal pathology (especially those who are diazoxide-unresponsive) and complements the standard NGS, which typically takes 4–6 weeks. This aids in early judicious nuclear imaging and surgical excision and prevents unnecessary prolongation of medical therapy [Table 1].[6] Conversely, an autosomal dominant or biallelic recessive mutation signifies diffuse pancreatic disease, obviating the need for further nuclear imaging.[7] However, due to the limited availability and high cost of rapid gene analysis in the local setting, the approach to a case of CHI rather depends on unresponsiveness to diazoxide therapy, which can indirectly indicate an underlying mutation in the K-ATP channel related genes.
| Pattern of inheritance | Zygosity | Histological type | Diazoxide responsiveness | Comment |
|---|---|---|---|---|
| AR | Biallelic (homozygous or compound heterozygous) | Diffuse | UR | Most require near total pancreatectomy |
| Monoallelic (father) | Focal (rarely diffuse) | UR |
Focal lesions- Loss of maternal 11p15 gene in a limited area of pancreatic tissue with concomitant paternal isodisomy Diffuse histology- Missed mutation in the deep intronic or regulatory region of a maternal gene, missed on routine Sanger sequencing. Focal lesionectomy- Curative in almost all patients |
|
| Monoallelic (mother) | Diffuse | UR | Spontaneous resolution with time; near total pancreatectomy can be averted | |
| AD | Heterozygous | Diffuse Atypical or LINE-HI |
Milder; usually responsive |
If K-ATP channel activity was completely impaired– maybe UR to diazoxide, needing octreotide therapy or near total pancreatectomy. Atypical or LINE-HI(scattered distribution of abnormal islet cells in the pancreas) can occur in certain AD somatic mutations of ABCC8, GCK and HK1genes. |
ABCC8: ATP-binding cassette subfamily C member 8, AD: autosomal dominant, AR: Autosomal recessive, GCK: Glucokinase, HK1: Hexokinase, K-ATP: Adenosine triphosphate sensitive potassium, LINE-HI: Localized islet nuclear enlargement, UR: Unresponsive
The various pharmacological treatments tried in CHI are shown in Table 2. Diazoxide forms the mainstay of medical therapy, and most responsive patients are well-controlled at a dose of <10 mg/kg/d.[8] Ideally, control of CHI should be ensured by subjecting the patient to safety fast. The duration of the fast varies depending on age, such that a neonate, an infant, and an older child should maintain BG levels >65 mg/dL after a fast of 6–12 h, 12–18 h, and 18 h. respectively.[6] A small percentage of diazoxide-unresponsive (UR) cases may respond to IV or SC octreotide [Table 2]. Children who respond to octreotide can be shifted to long-acting somatostatin analogs such as octreotide long-acting release (LAR) and lanreotide autogel. Although off-label, their use in patients with CHI has reported good control and improved quality of life.[9] Le Quan Sang et al. demonstrated a successful switch with octreotide LAR injections by overlapping the first two long-acting injections with daily SC injections.[10] Liver function tests and ultrasound for the appearance of gallstones should be performed periodically in children on long-acting octreotide [Table 2].[11] Trials with sirolimus and nifedipine in diazoxide UR patients have shown variable results.[12-14] At present, such therapy can be considered experimental, with no conclusive evidence advocating its use as standard therapy in patients with diazoxide-UR CHI.[15] In children with persistent hypoglycemia, despite all pharmacological measures and not being candidates for surgical therapy, continuous glucose intake, especially at night, can be provided through a nasogastric or gastrostomy tube. Uncooked cornstarch, a complex carbohydrate with a slow release of glucose, can be tried at 1–2 g/kg/d as a single bedtime dose or in divided doses throughout the day.
| Drug | Mechanism of action | Dose and frequency | Route | Adverse effects | Comments |
|---|---|---|---|---|---|
| Diazoxide | Binds to the K-ATP channel to keep it open | Starting dose 5 mg/kg/d; can titrate up to 15 mg/kg/d Every 8–12 h |
Oral |
Acute:Fluid retention, CCF, pericardial effusion, pulmonary hypertension, hyponatremia Chronic:Hypertrichosis, coarsening of facies, neutropenia, thrombocytopenia, hyperuricemia |
Concomitant hydrochlorothiazide - fluid overload and synergistically improves blood glucose control. CBC, uric acid at initiation and 6 monthly thereafter; baseline 2D-ECHO at initiation and after 1 week |
| Octreotide | Somatostatin receptor analog | 5–20 μg/kg/d; rarely up to 50 μg/kg/d Every 6–8 h |
SC or IV | Tachyphylaxis after initial good response. At high doses: Necrotizing enterocolitis, cholelithiasis, and growth failure |
An increment in the dose after a couple of days may be required. Growth monitoring, LFT and USG abdomen 6 monthly recommended |
| Long-acting Octreotide analogs (Octreotide LAR and Lanreotide autogel) | Somatostatin receptor analog | Cumulative dose of daily injections (Octreotide – LAR) or 30–60 mg of lanreotide once in 4–6 weeks | IM | Growth failure, elevated liver enzymes, gallstones | Growth monitoring, LFT and USG abdomen 6 monthly recommended |
| Glucagon | Glycogenolysis, gluconeogenesis, reduced hepatic uptake of glucose | In acute hypoglycemia: 30 µg/kg as a single dose or 2.5–20 µg/kg/h infusion to maximum dose of 0.5–1 mg |
IM, IV or SC | Vomiting, respiratory distress, necrolytic migratory erythema (rare) | IV line set to be changed every 12 hours. Continuous subcutaneous infusions like an insulin pump have been tried but are unreliable due to the precipitation of glucagon and infusion line blockage. |
| Sirolimus | mTOR inhibitor – beta cell proliferation, insulin secretion and induces insulin resistance | 0.5–1 mg/m2/d; dose can be titrated to achieve serum sirolimus concentration of 5–15 ng/mL; every 24 hours | Oral | Immunosuppression, increased infection and lymphoma risk; pneumonitis; hepatic and renal dysfunction; dyslipidemia | CBC, LFT, RFT, lipid levels. Watch for respiratory symptoms. |
CHI: Congenital hyperinsulinism of infancy, 2D-ECHO: 2-dimensional echocardiography, CBC: Complete blood count, CCF: Congestive cardiac failure, IM: Intramuscular, IV: Intravenous, K-ATP: Adenosine triphosphate sensitive potassium, LAR: Long-acting release, LFT: Liver function test, mTOR: Mammalian target of rapamycin, RFT: Renal function test, SC: Subcutaneous, USG: Ultrasonography
In the absence of rapid gene testing, nuclear imaging with 18-F-DOPA PET/CT is performed in children with poor response to medical therapy to rule out focal CHI (indicated by locally high standardised uptake value) with a sensitivity of 85% and positive predictive value of 96%.[16] A newer radiotracer compound, Gallium-68-NODAGA exendine-4 (which is a glucagon-like peptide-1 agonist), is emerging as an alternative to conventional 18F-DOPA PET/CT imaging, which requires a fasting state and encounters more frequent hypoglycemia.[17]
Detection of focal CHI allows lesionectomy and can result in a complete cure of CHI in 97% of cases.[18] However, at times, a lesion in the head of the pancreas, close to the bile duct insertion, may be difficult to excise, necessitating continuation of the conservative therapy. Children with diffuse CHI UR to medical therapy may benefit from a near total pancreatectomy in which 98% of the pancreas is removed, leaving a small rim of tissue surrounding the pancreatic and bile ducts. However, there is a growing preference for conservative management over surgical treatment in children with diffuse CHI, relatively euglycemic on medical therapy.[19] Further, a high possibility of post-surgical complications such as persistent hypoglycemia, diabetes mellitus, and exocrine pancreatic deficiency has been reported. Almost all children develop insulin-dependent diabetes by the second decade, and two-thirds require pancreatic enzyme supplementation on follow-up.[8] Irrespective of diazoxide-responsiveness, a genetic diagnosis can be made in 40–50% of patients with CHI. As per the recent international guidelines, genetic testing is recommended to be performed in all cases of suspected CHI who are diazoxide-responsive but continue to require therapy beyond 12 weeks of age.[7] Other than the K-ATP channel genes as described in Table 1, mutations in GCK can sometimes lead to severe diazoxide-UR CHI,[20]whereas mutations in GLUD1, HNF4A, HNF1A, HADH, and SLC16A1 cause diffuse but milder hyperinsulinism, responsive to medical therapy.[21] In those 50–60% of cases in whom no mutation is identified, CHI is usually diffuse, diazoxide-responsive, and remits with time.[6] In diazoxide UR infants, the chance of finding a positive mutation is almost 90%.[22]
Ambulatory management of CHI includes the timely administration of medications and maintaining a BG log through daily intermittent BG checks. A recent study revealed a diurnal pattern of hypoglycemia in children with CHI, occurring most frequently between 3 a.m. and 6 a.m. and pre-meal.[23] This could provide the basis for evaluating the effectiveness of self-monitoring of BG during specific times in the day when the child is at greatest risk of having a hypoglycemic episode.
Although the use of continuous glucose monitoring systems (CGMS) as a monitoring tool has been increasing, the accuracy of CGMS in patients with CHI has often been questioned. This is due to high variability in the setting of hypoglycemia, poor positive predictive value for low BG readings, and devices not being licensed for use in the very young. However, despite these drawbacks, it has been seen that CGMS aids in the detection of twice as many hypoglycemia events as compared to intermittent checks by glucometer.[24,25] The relationship between time in range (TIR) on a CGMS and neurodevelopmental outcome needs to be established.
The natural history of patients with CHI depends on the setting in which it occurs. Neonates born small for gestational age or with perinatal asphyxia and infants of diabetic mothers may present with transient hyperinsulinemic hypoglycemia. At times, they may require a short duration of diazoxide therapy, which can be tapered and stopped within 1 year of age. Genetic CHI secondary to HNF4A and HNF1A mutations is diazoxide-responsive and remits with time but causes the development of monogenic diabetes in the young subsequently.[4] Similarly, certain dominant ABCC8 mutations may also have a dual phenotype of CHI followed by early onset diabetes.[26] The CHI can last for a variable period and may even persist into adulthood. Interestingly, as was demonstrated in our case, adults with such mutations are at risk of developing early-onset diabetes even without features of CHI in the past.[27]
Neurodevelopmental affection in the form of behavioral issues, epilepsy, cognitive impairment, motor delay, learning disabilities, and speech delay has been observed in up to half of all children with CHI.[8] Presentation within the first week of life and the requirement of a high dose of diazoxide, irrespective of underlying genetic etiology and histopathological type, are predictors of adverse neurodevelopment.[28] It has been suggested that an early diagnosis and initiation of treatment can help prevent the development of neurological morbidities.[8]
Neonatal hypoglycemia can sometimes be the only manifestation of congenital hypopituitarism, like in our second case.[29] The incidence of congenital hypopituitarism varies between 1 in 4000 and 10,000.[30] The clinical presentation may vary greatly from no symptoms to non-specific symptoms with or without phenotypic clues.[30] Obvious phenotypical features of midline anomalies such as cleft lip and palate, single central incisor, micropenis, and divarication of the recti and radiological features such as agenesis of the corpus callosum and optic nerve hypoplasia can be pointers to hypopituitarism. Neonates with such phenotypic and radiological features should be evaluated for hypopituitarism despite no clinical features and need to be followed up regularly for evolving pituitary hormone defects.[31,32]
GH deficiency (GHD) is likely to present with neonatal hypoglycemia, especially when associated with multiple pituitary hormone deficiencies (MPHD).[33] GH and cortisol act synergistically to elevate BG, and replacement of both hormones is necessary to maintain euglycemia in children with hypopituitarism.[34] Cortisol and GH are predominantly catabolic hormones that initiate gluconeogenesis and lipolysis and provide substrates for metabolism. TSH deficiency may present as prolonged physiological jaundice and, rarely, hypoglycemia.[35]
A careful and detailed medical history that may predispose the child to congenital, acquired, or syndromic forms of hypopituitarism needs to be noted. Most of the neonates with congenital hypopituitarism are usually born with normal anthropometry at birth. Although up to 52% of patients with hypopituitarism may have postnatal complications such as hypoglycemia, hyponatremia, and recurrent sepsis, the diagnosis is made in the neonatal period in only 23%.[36] The presence of central hypothyroidism is a simple clue to the possibility of an MPHD and underlying malformation of the pituitary gland.[37]
Even though low GH and cortisol levels during hypoglycemia are not diagnostic, they are significant pointers to the possibility of hypopituitarism.[29] Due to the lack of circadian rhythm for cortisol secretion in the initial 6 months, random cortisol assessment may be useful to establish cortisol deficiency. Mehta et al., validated that the combination of 8 a.m. serum cortisol >175 nmol/L (6.34 µg/dL) and 30-min cortisol >540 nmol/L (19.56 µg/dL) post-ACTH stimulation test excludes congenital adrenal insufficiency with high specificity (100%) and sensitivity (80%).[36] False-negative results can occur even in infants with ACTH deficiency as placental corticotropin-releasing hormone continues to stimulate adrenals for a few weeks after birth.[29,36] However, cortisol values at either 30 or 60 min below 500 nmol/L (18 µg/dL) can establish adrenal insufficiency.[38]
A random GH concentration of ≤5 ng/mL (5 µg/L) during the first 7 days of life, accompanied by other pituitary hormone deficiencies and/or the MRI abnormality, is sufficient to diagnose GHD.[39,40] Binder et al. suggested that a GH cutoff of 7 µg/L as measured on a neonatal screening card by a highly sensitive polyclonal enzyme-linked immunosorbent assay gave 100% sensitivity and 98% specificity.[41] Physiological elevation of GH levels in the neonatal period, physiological decline in insulin-like growth factor-1 levels in the first 15– 18 months of life, inherent risks of GH stimulation tests in the neonatal period, and lack of specificity of low GH with hypoglycemia to diagnose GHD are important diagnostic challenges in the diagnosis of neonatal GHD.[38,39,41]
MRI of brain and pituitary gland is mandatory to look for midline brain abnormalities such as absent or hypoplastic corpus callosum, absent septum pellucidum, schizencephaly, heterotopia, and optic nerve hypoplasia; structural changes such as hypoplastic pituitary gland with ectopic PP or undescended PP, and an interrupted or hypoplastic pituitary stalk and Rathke’s cleft cysts.[42]
Several transcription factors have been closely associated with pituitary development and Rathke’s cleft cyst, of which IsL1 (Islet-1) deletion has 100% penetrance.[42] POU1FI (PIT-1), PROP1, HESX1, LHX3, LHX4, SOX2, SOX3, OTX2, TBX19 (T-PIT), DAX-1, etc., are some of the common genetic mutations associated with congenital hypopituitarism. LHX3 mutation is associated with deficiencies of GH, TSH, luteinizing hormone, and follicle-stimulating hormone, but rarely ACTH, and the PP is grossly unaffected.[43] Affected individuals have short, rigid cervical spine with limited neck rotation and trunk movement with sensorineural hearing loss. The pituitary morphology may vary from a small to an enlarged anterior pituitary with a lesion suggestive of a microadenoma.[44] Thus, genetic testing complements the clinical, biochemical and radiological diagnosis in children with hypopituitarism and helps to predict the evolution of the disease.[45,46]
Evaluation for cortisol deficiency and cortisol replacement in a dose of 9–12 mg/m2/d in three divided doses before thyroxine replacement is necessary to prevent adrenal crisis. Subsequently, thyroxine is initiated in a dose of 10–15 µg/kg/d if TSH deficiency exists. In our case, hydrocortisone was initiated, and subsequently, thyroxine was added. Supplementation with cortisol will increase the free water excretion and may unmask diabetes insipidus and should be screened for. GH treatment can be commenced during the neonatal period in neonates with persistent hypoglycemia (despite hydrocortisone supplementation) with daily SC recombinant human GH injections in a dose of 22–35 µg/kg/d in the evening to mimic physiological GH release.[39] Lower doses (10–20 µg/kg/d) can also lead to a comparable response in neonates.[30] A small dose of GH requirement in neonates resulted in challenges with the practical delivery of accurate dosing. Hence, the authors used alternative day GH dosing based on personal experience and preferences. There is a scarcity of data on GH use in neonates.
CONCLUSION
CHI and hypopituitarism are important causes of persistent and recurrent hypoglycemia in newborns, with significant neurological implications in cases of delayed diagnosis and treatment. A systematic focus on clinical, laboratory diagnostic pointers, and genetics would clinch the diagnosis leading to early therapy initiation. Management is challenging in the neonatal age group, and an individualized approach based on the response to medications, underlying genetic etiology, and radiological findings should be adopted. Further, data on optimal dosing and delivery of GH treatment in neonates and better treatment options in diazoxide-UR cases are needed from research and practical experience. With improved accessibility and affordability, appropriate use of genetic analysis tools helps in understanding disease progression, prognosis, precision medication, and finally, genetic counseling where indicated.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript, and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Neonatal identification of pituitary aplasia: A life-saving diagnosis. Review of five cases. Horm Res. 2004;62:10-6.
- [CrossRef] [Google Scholar]
- Recommendations from the pediatric endocrine society for evaluation and management of persistent hypoglycemia in neonates, infants, and children. J Pediatr. 2015;167:238-45.
- [CrossRef] [Google Scholar]
- Genetic characteristics of patients with congenital hyperinsulinism. Curr Opin Pediatr. 2018;30:568-75.
- [CrossRef] [Google Scholar]
- Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin Endocrinol (Oxf). 2013;78:803-13.
- [CrossRef] [Google Scholar]
- The contribution of rapid KATP channel gene mutation analysis to the clinical management of children with congenital hyperinsulinism. Eur J Endocrinol. 2011;164:733-40.
- [CrossRef] [Google Scholar]
- International guidelines for the diagnosis and management of hyperinsulinism. Horm Res Paediatr. 2024;97:279-98.
- [CrossRef] [Google Scholar]
- Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabet Med. 2019;36:9-21.
- [CrossRef] [Google Scholar]
- Treatment with long-acting lanreotide autogel in early infancy in patients with severe neonatal hyperinsulinism. Orphanet J Rare Dis. 2017;12:108.
- [CrossRef] [Google Scholar]
- Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol. 2012;166:333-9.
- [CrossRef] [Google Scholar]
- A multicenter experience with long-acting somatostatin analogues in patients with congenital hyperinsulinism. Horm Res Paediatr. 2018;89:82-9.
- [CrossRef] [Google Scholar]
- Assessment of nifedipine therapy in hyperinsulinemic hypoglycemia due to mutations in the ABCC8 gene. J Clin Endocrinol Metab. 2017;102:822-30.
- [CrossRef] [Google Scholar]
- Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N Engl J Med. 2014;370:1131-7.
- [CrossRef] [Google Scholar]
- mTOR inhibitors for the treatment of severe congenital hyperinsulinism: Perspectives on limited therapeutic success. J Clin Endocrinol Metab. 2016;101:4719-29.
- [CrossRef] [Google Scholar]
- Congenital hyperinsulinism: Diagnosis and treatment update. J Clin Res Pediatr Endocrinol. 2017;9:69-87.
- [CrossRef] [Google Scholar]
- Managing congenital hyperinsulinism: Improving outcomes with a multidisciplinary approach. Res Report Endocrine Dis. 2015;31:103-17.
- [CrossRef] [Google Scholar]
- (68)Ga-NODAGA-exendin-4 PET/CT improves the detection of focal congenital hyperinsulinism. J Nucl Med. 2022;63:310-5.
- [CrossRef] [Google Scholar]
- Surgical treatment of congenital hyperinsulinism: Results from 500 pancreatectomies in neonates and children. J Pediatr Surg. 2019;54:27-32.
- [CrossRef] [Google Scholar]
- Conservatively treated congenital hyperinsulinism (CHI) due to K-ATP channel gene mutations: Reducing severity over time. Orphanet J Rare Dis. 2016;11:163.
- [CrossRef] [Google Scholar]
- Genetics of congenital hyperinsulinemic hypoglycemia. Semin Pediatr Surg. 2011;20:13-7.
- [CrossRef] [Google Scholar]
- Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98:E355-63.
- [CrossRef] [Google Scholar]
- Complexities in the medical management of hypoglycaemia due to congenital hyperinsulinism. Clin Endocrinol (Oxf). 2020;92:387-95.
- [CrossRef] [Google Scholar]
- Continuous glucose monitoring systems: Are they useful for evaluating glycemic control in children with hyperinsulinism? Horm Res Paediatr. 2019;92:319-27.
- [CrossRef] [Google Scholar]
- Continuous flash glucose monitoring in children with congenital hyperinsulinism; first report on accuracy and patient experience. Int J Pediatr Endocrinol. 2018;2018:3.
- [CrossRef] [Google Scholar]
- Novel de novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes. 2008;57:1935-40.
- [CrossRef] [Google Scholar]
- Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011;54:2575-83.
- [CrossRef] [Google Scholar]
- Neurologic outcomes of 90 neonates and infants with persistent hyperinsulinemic hypoglycemia. Pediatrics. 2001;107:476-9.
- [CrossRef] [Google Scholar]
- Severe, persistent neonatal hypoglycemia as a presenting feature in patients with congenital hypopituitarism: A review of our case series. J Pediatr Endocrinol Metab. 2019;32:767-74.
- [CrossRef] [Google Scholar]
- Congenital hypopituitarism during the neonatal period: Epidemiology, pathogenesis, therapeutic options, and outcome. Front Pediatr. 2020;8:600962.
- [CrossRef] [Google Scholar]
- Neonatal Hypopituitarism: Approaches to diagnosis and Treatment. J Clin Res Pediatr Endocrinol. 2019;11:4-12.
- [CrossRef] [Google Scholar]
- Congenital adenohypophysis aplasia: Clinical features and analysis of the transcriptional factors for embryonic pituitary development. J Endocrinol Invest. 2006;29:208-13.
- [CrossRef] [Google Scholar]
- Hypoglycemia: A potent stimulus to secretion of growth hormone. Science. 1963;140:987-8.
- [CrossRef] [Google Scholar]
- Postnatal glucose homeostasis in late-preterm and term infants. Pediatrics. 2011;127:575-9.
- [CrossRef] [Google Scholar]
- The role of growth hormone and cortisone on glucose and gluconeogenic substrate regulation in fasted hypopituitary children. J Clin Endocrinol Metab. 1976;42:846-56.
- [CrossRef] [Google Scholar]
- An update on the biochemical diagnosis of congenital ACTH insufficiency. Clin Endocrinol (Oxf). 2005;62:307-14.
- [CrossRef] [Google Scholar]
- Neonatal detection of congenital hypothyroidism of central origin. J Clin Endocrinol Metab. 2005;90:3350-9.
- [CrossRef] [Google Scholar]
- Diagnosis and treatment of primary adrenal insufficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2016;101:364-89.
- [CrossRef] [Google Scholar]
- Growth hormone deficiency (GHD) from birth to 2 years of age: Diagnostic specifics of GHD during the early phase of life. Horm Res. 2003;60:2-9.
- [CrossRef] [Google Scholar]
- Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: Growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr. 2016;86:361-97.
- [CrossRef] [Google Scholar]
- Rational approach to the diagnosis of severe growth hormone deficiency in the newborn. J Clin Endocrinol Metab. 2010;95:2219-26.
- [CrossRef] [Google Scholar]
- Rathke's cleft-like cysts arise from Isl1 deletion in murine pituitary progenitors. J Clin Invest. 2020;130:4501-15.
- [CrossRef] [Google Scholar]
- Genetic forms of hypopituitarism and their manifestation in the neonatal period. Early Hum Dev. 2009;85:705-12.
- [CrossRef] [Google Scholar]
- Mechanisms underlying pituitary hypoplasia and failed cell specification in Lhx3-deficient mice. Dev Biol. 2008;313:118-29.
- [CrossRef] [Google Scholar]
- Hypopituitarism oddities: Congenital causes. Horm Res. 2007;68(Suppl 5):138-44.
- [CrossRef] [Google Scholar]
- Panhypopituitarism presents as life-threatening heart failure caused by an inherited microdeletion in 1q25, including LHX4. Pediatrics. 2012;129:e529-34.
- [CrossRef] [Google Scholar]




