
Translate this page into:
Neonatal diabetes mellitus with congenital hypothyroidism (NDH) syndrome caused by GLIS3 mutation: A case report and review of literature

*Corresponding author: Shaila Sanjay Pachapure, Department of Paediatrics, Jawaharlal Nehru Medical College, Belagavi, Karnataka, India. drshailams@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Pachapure SS, Badiger S, Tadakanahalli S, Franco ED, Manthale A, Kulkarni V. Neonatal diabetes mellitus with congenital hypothyroidism (NDH) syndrome caused by GLIS3 mutation: A case report and review of literature. J Pediatr Endocrinol Diabetes 2022;2:86-9.
Abstract
Neonatal diabetes mellitus with congenital hypothyroidism (NDH) syndrome (OMIM: 610199) caused by GLIS3 gene mutations is a multisystem disorder. To date, only 23 cases of NDH syndrome have been reported worldwide. We report a child diagnosed on day 24 of life with neonatal diabetes, congenital hypothyroidism, congenital glaucoma, and dysmorphic facial features. Genetic analysis revealed a homozygous pathogenic frameshift variant (p.Gln361Profs*193) in the GLIS3 gene, located on chromosome 9p24.2. Our report confirms that infants with neonatal diabetes and congenital hypothyroidism along with facial dysmorphism should be screened for GLIS3 mutations.
Keywords
Congenital hypothyroidism
GLIS3 mutation
Neonatal diabetes mellitus

INTRODUCTION
Neonatal diabetes mellitus (NDM) is defined as diabetes diagnosed within 6 months of age. It is a rare condition with an estimated incidence of one in 90,000–160,000 live births.[1] With advancement in molecular genetics, pathogenic variants in more than 30 genes have been found to cause neonatal diabetes.[2] Genetic mutations causing NDM may result in abnormal pancreatic development, β-cell function, or destruction, which cause either transient or permanent NDM.[1] Early diagnosis of neonatal diabetes helps predicting the clinical course and outcome.
The GLIS3 (Gli-similar 3) gene is located on chromosome 9p24.2 in humans. It has 11 exons, with only 2–10 being coding regions. The GLIS3 gene encodes a factor belonging to the family of Krüppel-like zinc finger transcription factors. The GLIS3 protein either represses or enhances the expression of various target genes in different organs including pancreas, thyroid, eyes, liver, and kidneys.[3] In the pancreas, GLIS3 has a critical role in β-cell development and its endocrine function. It also binds to the insulin (INS) promoter region and enhances its transcription.[4]
GLIS3 mutations causing NDH syndrome (autosomal recessive inheritance) were first reported in 2006, in a family from Saudi Arabia. Taha et al. in 2003 described two siblings who had intrauterine growth restriction (IUGR), NDM, congenital hypothyroidism, facial dysmorphism, congenital glaucoma, progressive hepatic fibrosis, and renal cystic dysplasia.[5] They expired in early childhood.
Three years later, Senee et al. found homozygous GLIS3 mutations as the cause of NDH syndrome in the original family and two other families from Saudi Arabia.[6] To date, 23 patients with GLIS3 pathogenic variants (homozygous or compound heterozygous) have been reported worldwide. (PMID: 35410112).
Herein, we present a case of neonatal diabetes caused by a GLIS3 mutation. This is, to the best of our knowledge, the first report of GLIS3-NDH from India.
CASE REPORT
A baby boy was brought to our hospital on day 11 of life, in view of poor weight gain and hyperglycemia. He was the second child of a second degree consanguineously married couple. Anomaly scan in third trimester revealed dilated left renal pelvis (14 mm) with a single umbilical artery. He was born at term (38 + 3 weeks) by vaginal delivery, with no neonatal asphyxia. Birth weight was 1.8 kg. As the mother had reduced milk secretion, diluted cow’s milk was started on day 4 of life at home. The baby became lethargic on day 5 of life, hence was admitted in a private hospital for the management of hyperglycemia (296 mg/dL).
At admission to our hospital, the baby was lethargic and dehydrated with normal vital signs. His weight was 1.68 kg, length was 43 cm, and head circumference was 33.5 cm. He had severe wasting, wide open anterior and posterior fontanels with corneal clouding, thin upper lip, long columella, large bulbous nose, and left cryptorchidism [Figure 1a]. Systemic examination was otherwise unremarkable. Random blood glucose reading was 195 mg/dL. Urine ketones were negative. Initial resuscitation was done with normal saline bolus 10 mL/kg, followed by maintenance fluids and first line antibiotics.
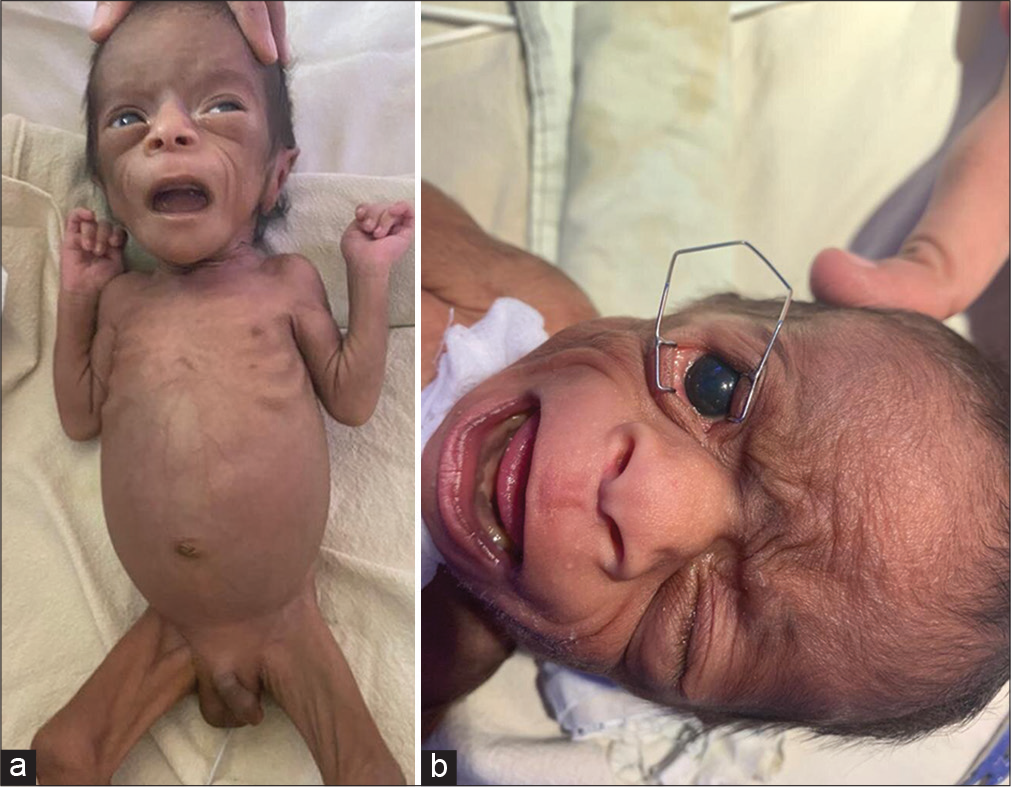
- (a) Patient with generalized wasting, with corneal clouding, thin upper lip, long columella, large bulbous nose, and left cryptorchidism. (b) Photograph of the face showing increased corneal diameter (megalocornea) (11 mm).
Laboratory investigations revealed negative septic screen with deranged renal functions (urea/creatinine – 68/1.1 mg/dL). Urine output improved with fluid replacement, but creatinine remained persistently high (ranging from 1.14–1.56 mg/dL). The liver function tests and serum electrolytes were normal. High blood glucose levels were noted only on four occasions during the initial 10 days of NICU stay (ranging from 140– 390 mg/dL), requiring subcutaneous regular insulin. After the baby was stabilized, breast milk and formula feeds were reinitiated, with blood glucose values remaining within normal limits (60–120 mg/dL).
Increased corneal diameter (megalocornea) (11 mm) [Figure 1b] and high intraocular pressure of 37 and 40 mm of Hg (iCare tonometer) were noted in the right and left eye, respectively. Hence, he was treated with timolol eye drops (0.25%).
Cranial ultrasonogram (USG) was normal. Abdominal USG revealed normal liver and bulky kidneys with mildly prominent left renal pelvis (6 mm). Thyroid screening was suggestive of congenital hypothyroidism, with high TSH (>150 mIU/L) (normal 1.19–10.72 mIU/L) and low free T4 of < 0.1 ng/dL (normal – 1.18–2.49 ng/dL).[7] Treatment was initiated with levothyroxine in a dose of 25 µg during hospital stay (15 µg/kg/day). After 2 weeks, the dose was increased to 37.5 µg (22 µg/kg/day) as repeat free T4 was still low (0.55 ng/dL) with TSH being 40 mIU/L.
On day 24 of life, blood glucose levels were persistently high; hence, injection glargine was started. HbA1C was noted to be 6.9% (HPLC Biorad 10). Maintaining euglycemia was a difficult task due to fluctuating hypoglycemia and hyperglycemia (GRBS ranging from 48 to >500 mg/dL). In view of the frequent hypoglycemia, the dosage interval of glargine was changed from 24 to 36 h.
Considering the classical features of persistent hyperglycemia with congenital hypothyroidism and congenital glaucoma, the diagnosis of NDM with congenital hypothyroidism (NDH) syndrome (secondary to GLIS3 mutation) was suspected, as other genes have not been reported for above association. After taking the informed consent from parents, genetic analysis was performed.
The baby was discharged home on day 40 of life with a weight of 1.755 kg, with insulin analog therapy and regular home blood glucose monitoring. Parents could not come for follow-up due to the COVID-19 pandemic. However, regular telephonic follow-up was done. At home, the mother had given glargine only on four occasions in a month, as the blood glucose levels were normal even without glargine. Hypoglycemic episodes were managed with parenteral correction once and later by oral glucose and breastfeeds at home. Later, the mother discontinued glargine due to frequent hypoglycemia.
At 3 months of age, he was taken to a nearby hospital for episodes of loose stools and hurried breathing when his weight was recorded as 1.9 kg. He succumbed to the illness after 2 days.
Genetic testing was performed by the Exeter Genomics Laboratory. DNA was extracted from blood samples and analyzed using a custom targeted next generation sequencing assay as previously described (ref PMID: 23771172). This assay allows analysis of the coding regions and conserved splice sites of the ABCC8, AGPAT2, BSCL2, CISD2, CNOT1, COQ2, COQ9, EIF2S3, EIF2AK3, FOXP3, GATA4, GATA6, GCK, GLIS3, HNF1B, IER3IP1, IL2RA, INS, INSR, KCNJ11, LPL, LRBA, MNX1, NEUROD1, NEUROG3, NKX2-2, PDX1, PTF1A, RFX6, SLC2A2, SLC19A2, STAT3, WFS1, and ZFP57 genes and can also detect partial/whole gene deletions and duplications. Variants are classified using the ACMG/AMP guidelines (Ellard et al. 2020 https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variantclassification-v4-01-2020.pdf; Richards et al 2015 PMID: 25741868).
The genetic analysis confirmed that the baby was homozygous for a pathogenic GLIS3 frameshift p.Gln361Profs*193 variant predicted to result in mRNA degradation through nonsense mediated decay. Both parents were heterozygous carriers. This result confirmed the diagnosis of NDH in this family (OMIM: 0610199).
DISCUSSION
Here, we describe the first Indian case with a homozygous mutation in the GLIS3 gene. The baby presented with NDM, congenital hypothyroidism, congenital glaucoma, and typical facial features. GLIS3 plays a key role in early embryogenesis and also cell regulation by functioning as repressor or activator of transcription.[8] Recently, the role of GLIS3 in type 1 and type 2 diabetes mellitus has also been investigated, providing new insights into pathogenesis.[9]
The onset of diabetes in NDH syndrome occurs within the first few weeks of life. Various case reports observed onset ranging from day 1 to 31 days of life. All patients required insulin. Fluctuating blood glucose levels were noted in our child similar to other reported cases.[10] The possible causes of hypoglycemia could be attributed to the role of GLIS3 in insulin secretion.[10]
Levothyroxine requirements are on the higher side in NDH syndrome than other causes of congenital hypothyroidism. Persistent high TSH has been noted in spite of high doses up to 20–35 µg/kg/day.[11,12] GLIS3 plays a role in the early stages of thyroid development from the pharyngeal endoderm and also in TSH receptor signaling and proliferation of follicles later.[3,13] Hence, NDH patients show varying thyroid anatomy on ultrasonography ranging from aplasia to normal size.[12,14,15]
Facial dysmorphism in our patient was similar to those reported by various authors and included low set ears, prominent eyes, epicanthal folds, flat nasal bridge, long philtrum, and thin upper lip.[10,11,16] The facial features were more evident with advancing age.[14]
Dimitri et al. reported osteopenia, thoracolumbar lordosis, bilateral sensorineural deafness, exocrine pancreatic dysfunction, hepatitis, hepatic fibrosis, and cirrhosis in various families further extending the clinical spectrum caused by GLIS3 mutations.[16,17] Exocrine pancreas functions were not evaluated in our patient due to financial constraints.
The varying phenotype is attributed to the tissue expression of variable length transcripts of the GLIS3 gene. Being a rare condition, further information is required for correlating genotype and phenotype variability. The cause for mortality in the previous case reports was pneumonia, respiratory failure, sepsis, and hepatic dysfunction. Few affected individuals are still alive, which may be explained by the absence of major organ involvement.
CONCLUSION
NDM is caused by single gene defects affecting the pancreatic β-cells and many systems. Identification of the genetic mutation, even in cases which cannot lead to effective treatment, guides us in finding other associated problems in syndromic cases and is important for genetic counseling. Further studies involving the role of GLIS3 in pancreas may help in therapeutic strategies.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Neonatal diabetes mellitus: An update on diagnosis and management. Clin Perinatol. 2018;45:41-59.
- [CrossRef] [PubMed] [Google Scholar]
- ISPAD clinical practice consensus guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19:47-63.
- [CrossRef] [PubMed] [Google Scholar]
- Transcription factor GLIS3: Critical roles in thyroid hormone biosynthesis, hypothyroidism, pancreatic beta cells and diabetes. Pharmacol Ther. 2020;215:107632.
- [CrossRef] [PubMed] [Google Scholar]
- The Krüppel-like zinc finger protein GLIS3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009;37:2529-38.
- [CrossRef] [PubMed] [Google Scholar]
- Neonatal diabetes mellitus, congenital hypothyroidism, hepatic fibrosis, polycystic kidneys, and congenital glaucoma: A new autosomal recessive syndrome? Am J Med Genet A. 2003;122A:269-73.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38:682-7.
- [CrossRef] [PubMed] [Google Scholar]
- Newborn screening guidelines for congenital hypothyroidism in India: Recommendations of the Indian society for pediatric and adolescent endocrinology (ISPAE)-Part I: Screening and confirmation of diagnosis. Indian J Pediatr. 2018;85:440-7.
- [CrossRef] [PubMed] [Google Scholar]
- GLIS3, a novel member of the GLIS3 subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003;31:5513-25.
- [CrossRef] [PubMed] [Google Scholar]
- Emerging roles of GLIS3 in neonatal diabetes, Type 1 and Type 2 diabetes. J Mol Endocrinol. 2017;58:R73-85.
- [CrossRef] [PubMed] [Google Scholar]
- Case report: Neonatal diabetes mellitus caused by a novel GLIS3 mutation in twins. Front Endocrinol (Lausanne). 2021;12:673755.
- [CrossRef] [PubMed] [Google Scholar]
- Case report: Extended clinical spectrum of the neonatal diabetes with congenital hypothyroidism syndrome. Front Endocrinol (Lausanne). 2021;12:665336.
- [CrossRef] [PubMed] [Google Scholar]
- Extended clinical features associated with novel GLIS3 mutation: A case report. BMC Endocr Disord. 2017;17:14.
- [CrossRef] [PubMed] [Google Scholar]
- GLIS3 as a critical regulator of thyroid primordium specification. Thyroid. 2020;30:277-89.
- [CrossRef] [PubMed] [Google Scholar]
- An emerging, recognizable facial phenotype in association with mutations in GLI-similar 3 (GLIS3) Am J Med Genet A. 2016;170:1918-23.
- [CrossRef] [PubMed] [Google Scholar]
- The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract Res Clin Endocrinol Metab. 2017;31:175-82.
- [CrossRef] [PubMed] [Google Scholar]
- Expanding the clinical spectrum associated with GLIS3 mutations. J Clin Endocrinol Metab. 2015;100:E1362-9.
- [CrossRef] [PubMed] [Google Scholar]
- Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol. 2011;164:437-43.
- [CrossRef] [PubMed] [Google Scholar]




