
Translate this page into:
Genetic diagnosis in XY disorders of sex development

*Corresponding author: Vandana Jain, Department of Pediatrics, Division of Pediatric Endocrinology, All India Institute of Medical Sciences, New Delhi, India. drvandanajain@aiims.edu
-
Received: ,
Accepted: ,
How to cite this article: Priyadarshini S, Sharma R, Jain V. Genetic diagnosis in XY disorders of sex development. J Pediatr Endocrinol Diabetes. 2024;4:58-69. doi: 10.25259/JPED_42_2024
Abstract
XY disorders of sex development (XY DSD) comprise a class of heterogeneous genetic entities that result in discrepancies between chromosomal, gonadal, and phenotypic sex due to a reduction in androgen synthesis or action. The chief categories of disorders include gonadal dysgenesis, disorders with reduced androgen production (biosynthetic defect – either alone or in conjunction with impaired glucocorticoid and/or mineralocorticoid synthesis), and insensitivity to androgen action. While conventional diagnostic modalities, encompassing karyotyping, biochemistry, radiology, and, in a few cases, diagnostic laparoscopy, help in formulating a provisional diagnosis, molecular genetic testing is key to arriving at a precise etiology. Besides ending the diagnostic uncertainty, a molecular diagnosis helps to predict the natural course in terms of pubertal development and potential for fertility, thus contributing to decisions on the gender of rearing; and guides on surveillance for extragenital features and the risk of recurrence in subsequent pregnancies. This paper broadly discusses the genetic basis of XY DSD, different modalities of genetic testing, and their utility and limitations.
Keywords
XY differences in sex development
46,XY differences in sex development
Gonadal dysgenesis
Testosterone biosynthetic defects
Molecular diagnosis of differences in sex development

INTRODUCTION
Disorders of sex development (DSD) are defined as congenital conditions in which the development of chromosomal, gonadal, and anatomic sex is atypical.[1] The incidence of DSDs is 1 in 4500–1 in 5500 live births.[2] Based on chromosomal constitution, DSDs are classified into XX, XY, and sex chromosomal DSDs. A detailed history, thorough physical examination, and tiered investigations help inform the type of DSD, the confirmation of which requires a karyotype prepared from peripheral blood lymphocytes.[3]
XX DSDs occur due to the virilization of female fetuses from defects in adrenal steroid biosynthetic pathways with increased androgen production (congenital adrenal hyperplasia [CAH]), maternal exposure to excess androgens ([exogenous androgen intake, maternal virilizing tumors], or the presence of testicular tissue [testicular DSD, ovotesticular DSD]). XY DSDs occur due to under-masculinization of male fetuses from defects in adrenal and/or gonadal steroid biosynthetic pathways [Figure 1], defective testicular development in utero (gonadal dysgenesis) [Figure 2], defects of the hypothalamic-pituitary-gonadal axis (hypogonadotropic hypogonadism), or androgen receptor (AR) defects; all of which lead to reduced androgen production or action. Sex chromosomal DSDs occur as a result of aberration from the normal sex chromosome complement, for example, mixed gonadal dysgenesis 45,X/46,XY and 46,XX/46,XY, and Klinefelter syndrome 47,XXY. The present review focuses on the genetics of XY DSD.
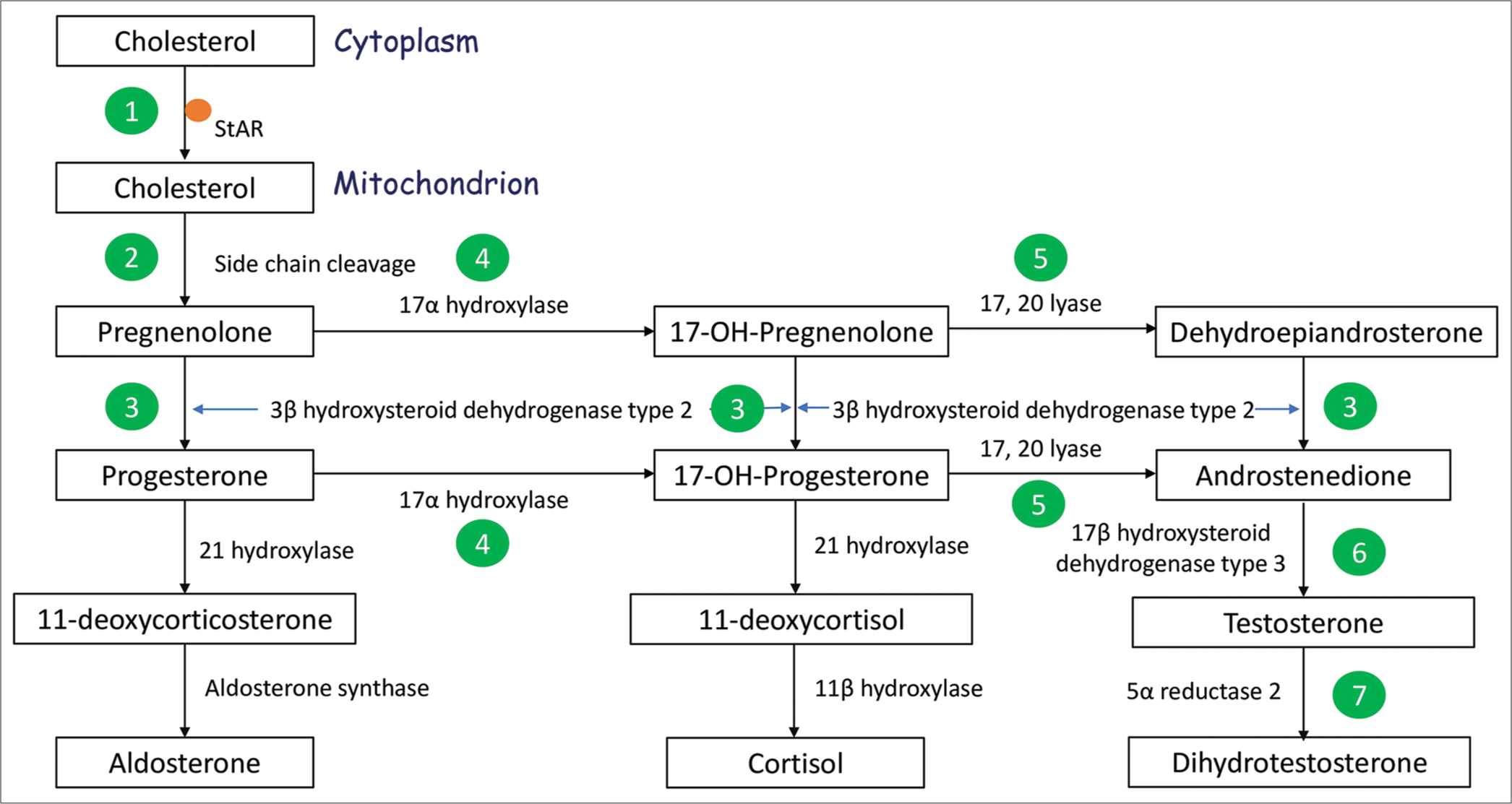
- Steroid biosynthetic pathway operating in the adrenal cortex and gonad. XY DSD due to impaired steroid biosynthesis can result due to defects in StAR encoding Steroidogenic Acute Regulatory protein (1), CYP11A1 encoding Side Chain Cleavage enzyme (2), and HSD3B2 encoding 3β hydroxysteroid dehydrogenase type 2 (3), all three of which result in mineralocorticoid and glucocorticoid insufficiency; due to defects in CYP17A1 encoding 17α hydroxylase (4) which result in mineralocorticoid excess and glucocorticoid insufficiency; and due to defects in CYB5A encoding cytochrome B5 type A (a co-factor for 17, 20 lyase enzyme) (5), HSD17B3 encoding 17β hydroxysteroid dehydrogenase type 3 (6), and SRD5A2 encoding 5α reductase type 2 (7), all three of which result in isolated XY DSD.
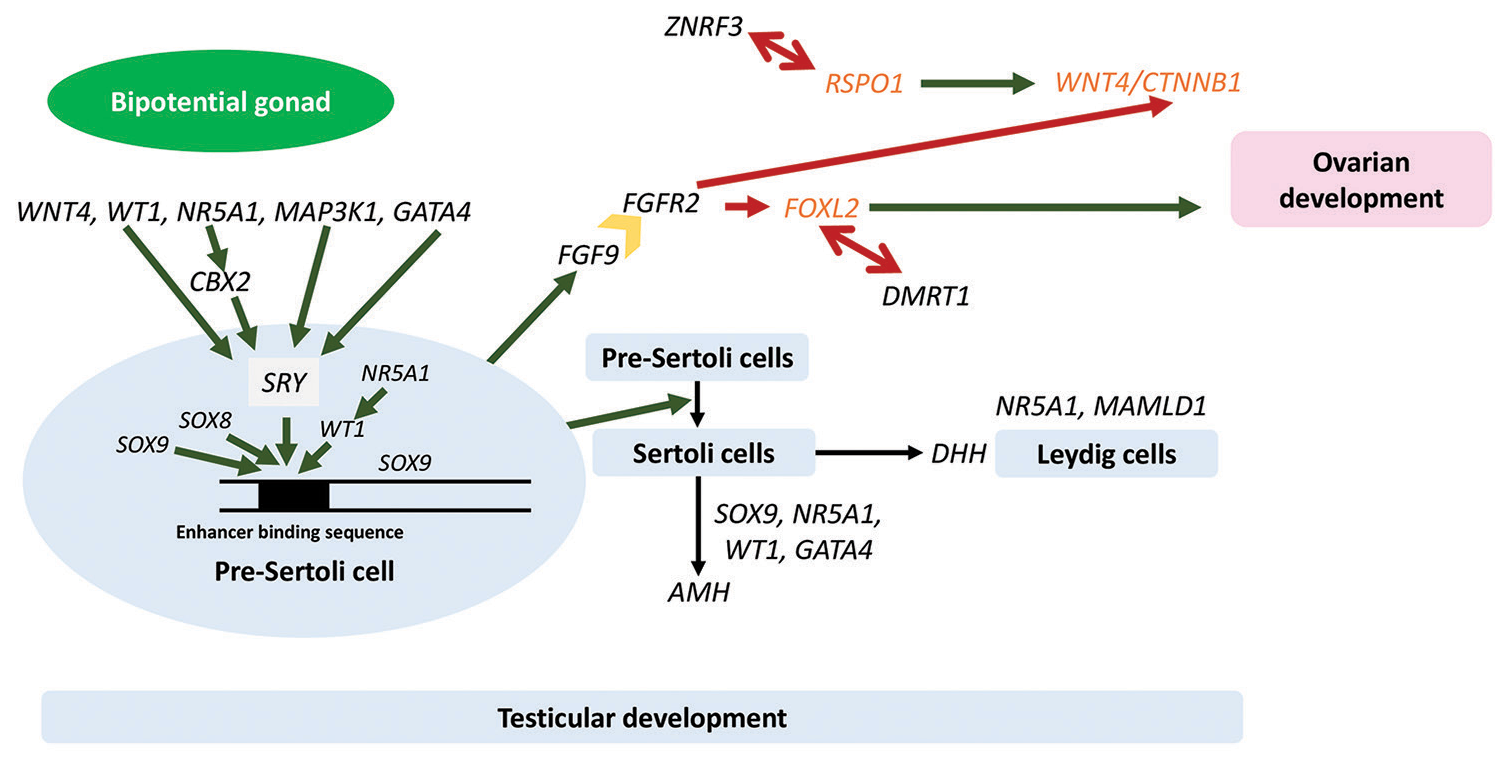
- Pathways of gonadal development. Dark green single arrows depict stimulatory pathways, dark red single arrows depict inhibitory pathways, dark red double arrows depict antagonistic relationships between protestes and pro-ovarian genes, and dark yellow block arrow depicts the protein product of FGFR2, the receptor for FGF9, a protestis protein which inhibits ovarian pathways after binding to its receptor, FGFR2. Genes and proteins in black are pro-testes, and genes and proteins in orange are pro-ovarian. WNT4: Wingless-type MMTV integration site family member 4, WT1: Wilms’ tumor 1, NR5A1: Nuclear receptor subfamily 5 group A member 1, MAP3K1: Mitogen-activated kinase kinase kinase 1, GATA4: GATA binding protein 4, CBX2: Chromobox 2, SOX9: SRYbox transcription factor 9, SOX8: SRY-box transcription factor 8, SRY: Sex-determining region Y gene, FGF9: Fibroblast growth factor 9, FGFR2: Fibroblast growth factor receptor 2, ZNRF3: Zing ring finger 3, DMRT1: Doublesex and mab-3 related transcription factor 1, MAMLD1: Mastermind like domain containing 1, DHH: Desert hedgehog, AMH: Anti-Müllerian hormone, RSPO1: R-spondin1, CTNNB1: Catenin-beta 1, FOXL2: Forkhead box L2.
CASE SCENARIOS
Case 1
A 2-month-old baby was brought to the pediatric outpatient with the concern of atypical genitalia. The gender assigned at birth was male. The parents felt that the baby’s phallus was very small, and he passed urine from an opening located at the base of the phallus. The baby was the first offspring of a non-consanguineously married couple, with a birth weight of 2.7 kg and an uneventful perinatal course. He had been feeding well and gaining weight appropriately. There was no history of recurrent vomiting, progressive hyperpigmentation, or a history of ambiguous genitalia or infertility in the family. On examination, vital parameters and anthropometry were normal, with no signs of dehydration or hyperpigmentation, and normal systemic examination. Scrotum was well-formed and rugose with intrascrotal gonads, and phallic length was 1 cm with chordee and penoscrotal hypospadias. Preliminary investigations yielded normal electrolytes and glucose, cortisol 17.1 µg/dL (reference 2.8–23), luteinizing hormone (LH) 8.4 IU/L (reference range 0.6–4.1), follicle-stimulating hormone (FSH) 4.4 IU/L (reference range 0.4–3.0), 17α-hydroxyprogesterone (17OHP) <0.05 ng/mL (Reference 0.11–1.7), testosterone 12 ng/dL (reference range 20–219), and anti-Müllerian hormone (AMH) 120 ng/mL (reference range 55–210). There were no müllerian structures on ultrasonography. Karyotype was 46,XY. Human chorionic gonadotropin (hCG) stimulation test was done, on which serum testosterone showed an inadequate rise to 59 ng/dL (reference >100) with a testosterone to androstenedione ratio of 0.5 (reference >0.8). The overall clinical picture (palpable gonads; absent Müllerian structures on ultrasonography; normal blood pressure, serum electrolytes, glucose, and cortisol; with low baseline and stimulated testosterone; and normal AMH) was in keeping with a diagnosis of testosterone biosynthetic defect without adrenal insufficiency, specifically 17β-hydroxysteroid dehydrogenase-3 deficiency.
The baby was prescribed three doses of Inj. testosterone 25 mg intramuscularly (IM) at monthly intervals, and subsequently underwent uneventful chordee correction and urethroplasty at the ages of 1 year and 2.5 years, respectively. At the age of 2 years and 8 months, however, he was brought to the pediatric emergency with fever, diarrhea, vomiting, and seizures. On examination, he appeared dehydrated and hyperpigmented. Investigations revealed sodium of 123 mEq/L, potassium 6.7 mEq/L, random blood glucose 25 mg/dL, cortisol 3.3 µg/dL, adrenocorticotropic hormone (ACTH) 1250 pg/mL (reference range 15–65), and 17OHP 0.3 ng/mL (Reference range 0.11–1.7). The diagnosis was revised to a salt wasting form of CAH such as 3β-hydroxysteroid dehydrogenase-2 deficiency, steroidogenic acute regulatory (StAR) defect, or side chain cleavage (CYP11A1) defect [Figure 1]. Dehydroepiandrosterone sulfate (DHEAS) was 0.46 µg/dL (reference 7–126). Exome sequencing identified the following [Box 1]:
| Gene | Exon | cDNA change | Protein change | Zygosity | Pathogenicity |
|---|---|---|---|---|---|
| StAR | 3 5 |
c. 297delG c. 562C>T |
(p.Ser100 ValfsTer9) (p.Arg188Cys) |
Compound heterozygous | Likely pathogenic Pathogenic |
StAR: Steroidogenic acute regulatory
A diagnosis of XY DSD and primary adrenal insufficiency due to StAR defect was made.
Learning point
Steroidogenic acute regulatory (StAR) and CYP11A1 defects can have a spectrum of presentation. The classical textbook description of presentation with salt wasting crisis or hypoglycemic seizures in the neonatal period, with near complete feminization of genitalia in an infant with XY DSD, is not always present. A molecular diagnosis made in advance could have averted a life-threatening adrenal crisis in our patient.
Case 2
A 2-month-old baby was brought to the pediatric outpatient with the chief complaint of atypical genitalia. The gender assigned at birth was male. Birth, development, and family history were non-contributory. On examination, the left testis was palpable in the inguinal region, while the right testis could not be palpated, and there was micropenis with a phallic length of 1.5 cm, chordee, and penoscrotal hypospadias (external masculinization score 4/12). The rest of the general physical and systemic examination was normal. On evaluation, the baby had a karyotype of 46,XY. Sonogram of the pelvis identified a uterus and both gonads in the inguinal canals. Biochemical investigations were as follows: LH 7.3 IU/L (reference range 0.6–4.1), FSH 6.7 IU/L (reference range 0.4–3.0), testosterone 214 ng/dL (reference range 20–219), and AMH 15.6 ng/mL (reference range 55–210). A diagnosis of XY DSD due to partial gonadal dysgenesis was made. Urine routine examination and ultrasonography of kidneys were normal. The molecular genetic testing was reported as follows [>Box 2]:
| Gene | Intron | cDNA change | Protein change | Zygosity | Pathogenicity |
|---|---|---|---|---|---|
| WT1 | 7 | c. 1264+1G>A | 5’ splice site | Heterozygous | Uncertain significance |
WT1: Wilms’ tumor 1
A provisional diagnosis of Denys–Drash syndrome was made. A surveillance plan to monitor for the development of Wilms’ tumor and nephrotic syndrome was devised, and parents were counseled about this. After 2 months, the mother presented to us with the concern of having noted an abdominal mass, which was identified on the sonogram as a well-circumscribed hypoechoic mass measuring 6.5 × 6.3 cm and arising from the lower pole of the left kidney. Detailed workup did not identify any local invasion or metastasis. A diagnosis of Wilms’ tumor was made, and the baby received neoadjuvant chemotherapy and underwent a left radical nephrectomy. He has been doing well on follow-up, with the plan to complete his course of adjuvant chemotherapy and monitor for the development of nephrotic syndrome or Wilms’ tumor in the contralateral kidney.
Learning point
A molecular diagnosis enabled the prediction and early identification of an otherwise aggressive tumor, at a stage when cure was likely.
GENETICS OF XY DSD
As opposed to XX DSDs, in which the vast majority of cases occur due to CAH, whose diagnosis rests on biochemical parameters, XY DSD is a highly heterogeneous group encompassing an array of genotypes and widely overlapping phenotypes. The final common pathway operates through a reduced androgen effect and can have manifestations ranging from mild under-masculinization (micropenis) through genital ambiguity to completely feminine external genitalia. XY DSD is broadly categorized into the following groups.
Disorders of gonadal development
Defects in genes that participate in normal testicular development can result in complete or partial gonadal dysgenesis.[4] Figure 2 depicts the pathway of gonadal development in males and females. Discussing briefly, the bipotential gonad develops from the embryonic mesoderm at about 4 weeks from conception. The development is mediated and sustained by genes such as Wilms’ tumor 1 (WT1), nuclear receptor subfamily 5 group A member 1 (NR5A1), and Wingless-type MMTV integration site family member 4 (WNT4). In fetuses with a Y chromosome, the sex-determining region Y (SRY) gene is activated by WT1, mitogen-activated kinase kinase kinase 1 (MAP3K1), GATA binding protein 4 (GATA4), and Chromobox 2 (CBX2). The protein product of SRY, along with the products of other genes such as NR5A1, WT1, and SRY-box transcription factor 8 (SOX8), in turn, increases the transcription of SRYbox transcription factor 9 (SOX9) by interacting with its upstream enhancer. SOX9 protein causes differentiation of pre-Sertoli cells into Sertoli cells and enhances transcription of fibroblast growth factor 9, which inhibits pathways that lead to ovarian development. Sertoli cells secrete AMH and desert hedgehog (DHH) homolog. AMH causes regression of müllerian structures such that fallopian tubes, uterus, cervix, and the upper one-third of the vagina are not formed, while DHH, along with SF1 (the protein product of NR5A1) and the protein product of Mastermind-like domain containing 1 (MAMLD1), promotes the development of Leydig cells. Leydig cells secrete testosterone and insulin-like peptide-3, both of which participate in testicular descent. A number of testes determining transcription factors encoded by genes such as SOX9, Doublesex, and mab-3 related transcription factor 1 (DMRT1) and Zing ring finger 3 (ZNRF3) repress ovarian development pathways that involve protein products of R-spondin1 (RSPO1), WNT4, Catenin-beta 1 (CTNNB1), Forkhead box L2 (FOXL2), and DAX1 (NR0B1).[5] It is worthwhile to note that the majority of the sex-determining genes are dosage-sensitive. Insufficiency of the pro-testes genes (WT1, NR5A1, GATA4, MAP3K1, CBX2, SOX9, SOX8, SRY, DHH, DMRT1, ZNRF3, and MAMLD1) and/or excess of the pro-ovarian genes (DAX1, WNT4, FOXL2, RSPO1, and CTNNB1) can cause impaired gonadal development and gonadal dysgenesis in individuals with XY karyotype.
Some of these molecular defects present with isolated gonadal dysgenesis, while others may have syndromic presentation, as they regulate the development of other organ systems as well. Key examples of the latter include Frasier syndrome (focal segmental glomerulosclerosis and gonadoblastoma) and Denys–Drash syndrome (diffuse mesangial sclerosis and Wilms’ tumor) due to defects in WT1 gene, congenital heart disease in GATA4 defects, primary adrenal insufficiency in NR5A1 defects, campomelic dysplasia in SOX9 defects, and polyneuropathy in DHH defects. Again, the presentation is variable in these, with the spectrum of presentations evolving over time as more and more patients are offered molecular testing. In individuals with XY DSD due to WT1 defects, up to 85% of individuals have renal pathology in the form of either early- or late-onset glomerulopathy or Wilms’ tumor.[6] For some of the variants, the genotype-phenotype correlation has been very strongly established. For example, missense variants in exons 8 and 9 of WT1 are associated with Denys– Drash syndrome, while splice site variants in intron 9 result in Frasier syndrome.[6,7] In defects of the NR5A1 (formerly SF1) gene, which has emerged as one of the commoner causes of XY DSD, the majority of patients present with a phenotype of partial gonadal dysgenesis with no adrenal insufficiency.
Disorders of gonadal development arise secondary to both single-nucleotide variants (SNVs), which are amenable to detection by gene sequencing-based methods (single gene and panel testing, whole exome sequencing [WES]), as well as copy number variants (CNVs), i.e., deletions or duplications, that are amenable to detection by chromosomal microarray (CMA)-based methods. Certain genes are described to harbor only SNVs, others are described to harbor only CNVs, and yet others are described to harbor a combination of both.[8]
Disorders of androgen biosynthesis
The classical pathway of androgen biosynthesis starts with cholesterol and cruises through a number of steps before culminating in the production of androgens [Figure 1]. Leydig cells produce testosterone, which is activated by 5α-reductase type 2 to dihydrotestosterone (DHT) in target tissues. Both testosterone and DHT act on AR and result in masculinization of the internal and external genitalia. Besides the classical pathway of androgen biosynthesis, an alternative pathway, also known as the “backdoor” pathway, operates for the synthesis of DHT from cholesterol that does not involve DHEA, androstenedione, and testosterone. In the backdoor pathway, 5α-reductase type 1 (SRD5A1) acts on 17OHP and progesterone. In addition, other enzymes such as aldo-keto reductases (AKR1C2/4), 17β-HSD5 (AKR1C3), and 17β-HSD6 (also called retinol dehydrogenase, RoDH) (HSD17B6) are critical mediators of this pathway. The backdoor pathway plays a crucial role in early male sexual development and physiological male mini-puberty during infancy. Defects in the genes AKR1C2/AKR1C4 result in XY DSD.[9] Androgen biosynthetic defects are inherited in an autosomal recessive fashion and arise as a result of SNVs that are amenable to genetic diagnosis by sequencing-based methods.[8] Based on their location in the classical pathway of steroid biosynthesis, the defects may present as isolated DSD (CYB5A, HSD17B3, 5α reductase [SRD5A2], and non-classical form of HSD3B2 defect), as DSD with combined mineralocorticoid and glucocorticoid insufficiency (StAR, CYP11A1, and HSD3B2), or as DSD with mineralocorticoid excess and glucocorticoid insufficiency (CYP17A1, and POR)[10,11] [Figure 1]. Defects in LH/choriogonadotropin receptor (LHCGR), a gene encoding receptor for both LH and hCG, cause impaired downstream signaling inside Leydig cells and result in reduced androgen synthesis.
Disorders of androgen action
Androgen action defect due to variation in the AR gene, located on the X chromosome, may occur due to both SNVs and CNVs (chiefly deletions).
Disorders of hypothalamic-pituitary-gonadal axis
Hypogonadotropic hypogonadism, identified chiefly in children presenting with delayed puberty, may also manifest in the neonatal period as genital ambiguity with micropenis and bilateral cryptorchidism. It occurs due to defects in genes that participate in development and migration of gonadotropin-releasing hormone (GnRH) neurons (Kallmann syndrome 1 sequence, Fibroblast growth factor 8, Fibroblast growth factor receptor 1, Prokineticin 2, Prokineticin receptor 2, and Chromodomain helicase DNA binding protein 7), GnRH secretion (KiSS-1 metastasis suppressor, Kisspeptin receptor, Tachykinin precursor 3, and Tachykinin receptor 3), and GnRH action (Gonadotropin-releasing hormone receptor).[12] Up to 60% of patients with idiopathic hypogonadotropic hypogonadism, chiefly due to defective migration of GnRH neurons, also have associated anosmia/hyposmia resulting in Kallmann syndrome. In general, the defects of the hypothalamic-pituitary-gonadal axis occur due to SNVs and can be identified using sequencing-based methods.[8]
Table 1 lists the genes implicated in various etiologies of XY DSD.
| Disease | Genes |
|---|---|
| Disorders of gonadal development | |
| Complete/partial gonadal dysgenesis | SRY, SOX9, WT1, GATA4, NR5A1, MAP3K1, DHH, MAMLD1, ZNRF3, DMRT1, CBX2, DHX37, DAX1, SOX8, WNT4 |
| Testicular regression syndrome | DHX37 |
| Ovotesticular DSD | NR5A1, DAX1, DMRT1, SRY |
| Disorders of androgen synthesis/action | |
| Disorders of androgen biosynthesis with adrenal insufficiency | DHCR7, StAR, CYP11A1, CYP17A1, HSD3B2, POR |
| Disorders of androgen biosynthesis without adrenal insufficiency | LHCGR, CYB5A, HSD17B3, SRD5A2, AKR1C2, AKR1C4 |
| Disorders of androgen action | AR |
| Hypogonadotropic hypogonadism | |
| Disorders of GnRH neuron development and migration | KAL1, FGF8, FGFR1, PROK2, PROKR2, CHD7 |
| Disorders of GnRH secretion | LEP, LEPR, KISS1, KISS1R, TAC3, TACR3, PCSK1 |
| Disorders of GnRH action | GNRHR |
SRY: Sex-determining region Y gene, SOX9: SRY-box transcription factor 9, WT1: Wilms’ tumour 1, GATA4: GATA binding protein 4, NR5A1: Nuclear receptor subfamily 5 group A member 1, MAP3K1: Mitogen-activated kinase kinase kinase 1, DHH: Desert hedgehog, MAMLD1: Mastermind like domain containing 1, ZNRF3: Zing ring finger 3, DMRT1: Doublesex and mab-3 related transcription factor 1, CBX2: Chromobox 2, DHX37: DEAH-box helicase 37, DAX1: Dosage-sensitive sex reversal, adrenal hypoplasia critical region, SOX8: SRY-box transcription factor 8, WNT4: Wingless-type MMTV integration site family member 4, DHCR7: 7-dehydrocholesterol reductase, StAR: Steroidogenic acute regulatory, CYP11A1: Side chain cleavage, CYP17A1: 17αhydroxylase, HSD3B2: 3βhydroxysteroid dehydrogenase type 2, SRD5A2: 5αreductase, AKR1C2: Aldo-keto reductase family 1 member C2, AKR1C4: Aldo-ketoreductase family 1 member C4, AR: Androgen receptor, KAL1: Kallmann syndrome 1 sequence, FGF8: Fibroblast growth factor 8, FGFR1: Fibroblast growth factor receptor 1, PROK2: Prokineticin 2, PROKR2: Prokineticin receptor 2, CHD7: Chromodomain helicase DNA binding protein 7, LEP: Leptin, LEPR: Leptin receptor, KISS1: KiSS-1 metastasis suppressor, KISS1R: Kisspeptin receptor, TAC3: Tachykinin precursor 3, TAC3R: Tachykinin receptor 3, PCSK1: Proprotein convertase subtilisin/kexin type 1, GNRHR: Gonadotropin releasing hormone receptor, LHCGR: Luteinizing hormone/choriogonadotropin receptor
CONVENTIONAL DIAGNOSTIC ALGORITHMS FOR EVALUATION OF XY DSD
The evaluation of a child with atypical genitalia begins with a complete medical history, detailed physical examination, and documentation of the findings systematically using the external genitalia score.[13] In an individual with XY karyotype, the evaluation is directed at identifying the presence of and localizing testicular tissue, establishing functionality of the testicular tissue, and assessing the ability of the testes to produce testosterone with its successful activation to DHT.[14]
Imaging
Imaging helps to localize gonads and assess the anatomy of the internal genitalia. The presence of testes and the absence of müllerian derivatives point to XY DSD due to androgen synthesis/action defect, whereas the presence/absence of gonads and the presence of müllerian derivatives point to XY DSD due to gonadal dysgenesis. Ultrasonography is the first line of imaging investigation, supplemented by a genitogram and/or magnetic resonance imaging (MRI) of the abdomen and pelvis, as needed.
Hormonal evaluation
A low level of testosterone during mini-puberty or puberty suggests either gonadal dysgenesis or biosynthetic defect. Serum AMH, a marker of immature Sertoli cell function, helps to distinguish isolated biosynthetic defect (normal AMH) from gonadal dysgenesis (low AMH). A normal AMH level is indicative of a functioning testicular tissue and is useful in the evaluation of patients with bilateral cryptorchidism, ovotestes, and XX testicular DSD. AMH level starts to rise soon after birth as Sertoli cells begin to increase, reaches a peak at 6 months of age, and gradually declines through childhood, becoming low during puberty.[15]
In addition to normal AMH and low testosterone, a low testosterone to androstenedione ratio (cut off of 0.8) is also suggestive of testosterone biosynthetic defect. When a biosynthetic defect is suspected, further evaluation is done to assess if the defect involves the glucocorticoid and/or mineralocorticoid pathways as well by measuring levels of plasma glucose, serum electrolytes (Na+/K+), serum cortisol, plasma ACTH, and when required, a steroid panel comprising 17OHP, 11-deoxycorticosterone, and DHEAS. When the suspicion of adrenal insufficiency is high with non-specific findings at baseline, dynamic testing using standard dose synacthen (ACTH) stimulation may be required.
If testosterone levels during mini-puberty or puberty are normal to high, 5α-reductase deficiency and androgen insensitivity syndrome (AIS) remain the chief possibilities. The testosterone to DHT ratio is commonly employed to differentiate between the two (high [>20] in 5α-reductase deficiency and low [<20] in AIS). However, the test lacks specificity, with elevated ratios noted in a major proportion of individuals with AIS.[16]
Evaluation of a prepubertal child with low levels of LH and FSH is challenging because quiescence of the hypothalamic-pituitary-gonadal axis results in investigations that are non-informative. This warrants activation of the androgen biosynthetic pathway by administering hCG, which is also an agonist at the receptor for LH (LHCG receptor). The usual protocol comprises IM administration of Inj. hCG 500–1500 IU/m2 every 48 h with an assessment of steroid profile 24 h after the last dose, but the protocol may have to be extended for up to 3 weeks in prolonged hCG testing. The latter is usually done when levels of both testosterone and its precursors are low after a standard hCG stimulation test.[17]
Surgery
Diagnostic laparoscopy is done if the gonads and/or Müllerian structures cannot be localized on routine imaging. It is typically combined with gonadal biopsy and/or gonadopexy.
Genetic studies
Karyotyping is an essential investigation in children with DSD. Other modalities, such as fluorescent in situ hybridization (FISH) and quantitative fluorescent–polymerase chain reaction (QF-PCR) to identify Y chromosome material, may be used in the interim due to their faster turnaround time. This is especially helpful in an infant with atypical genitalia in whom the diagnostic process needs to be expedited for sex assignment and alleviation of parental anxiety.
PITFALLS OF THE CURRENT DIAGNOSTIC ALGORITHMS
The major drawbacks of the current diagnostic protocol are as follows:
This requires a combination of tests, usually done in a stepwise manner, to be able to arrive at a specific diagnosis. This approach not only increases the time to reach an accurate diagnosis but also adds to the cumulative cost of testing.
Some of the tests do involve procedural difficulties, such as three IM injections and two sets of venous sampling for the hCG stimulation test, catheterization and radiation exposure for genitogram, and anesthesia or sedation for MRI.
Immunoassays have low specificity for steroid metabolites, while liquid chromatography-mass spectrometry/mass spectrometry is not routinely available and is costlier.
Irrespective of the technique employed, levels and ratios of various metabolites may overlap for different etiologies and are often non-pathognomonic. For instance, NR5A1 defects may present with a biochemical picture of gonadal dysgenesis, isolated testosterone biosynthetic defect, or with intact testosterone synthesis.[14] As already discussed, the commonly used testosterone to DHT ratio lacks specificity for diagnosing 5α-reductase deficiency.[16]
CASE FOR A GENETIC DIAGNOSIS IN XY DSD
Establishing a precise etiology
Molecular diagnosis ends the diagnostic odyssey for the family and the clinician. For the patient and their family, it leads to an acceptance of the diagnosis, brings about an understanding of “why this happened,” and thus helps to ease feelings of stigma, guilt, shame, and blame that often accompany a diagnosis of DSD. More importantly, it helps in bringing out the entity from under the blanket or umbrella term of DSD and gives an exact diagnosis to the child for the under or over-virilization, akin to other endocrinological conditions.
Predicting disease course/trajectory
A molecular diagnosis informs the clinician of hormone production and responsiveness, which can help in choosing appropriate androgen replacement therapy. For example, children with testosterone biosynthetic defects respond well to testosterone, whereas those with 5α-reductase type 2 deficiency and partial AIS (PAIS) benefit from the application of DHT. Molecular diagnosis also provides guidance regarding the most appropriate sex of rearing based on the data available in the literature on pubertal development, adult gender identity, sexual function, and fertility. However, as of now, such information is scant for several molecular etiologies, barring the most common ones, such as AIS, SRD5A2 defect, and HSD17B3 defect for which Sanger sequencing has been in use for some time. This literature is also mostly from Europe and the Middle East. With greater availability of molecular diagnoses using next-generation sequencing (NGS), the phenotypic spectrum, as well as the natural course of several molecular etiologies, will now attain greater clarity and will have the potential to guide future clinicians and families on optimum care.
Prediction of extragenital features
As highlighted in the section on types of XY DSD, a genetic diagnosis is key to predicting the onset of extragenital features. This enables putting in place a surveillance plan for the patient, which can potentially avert life-threatening complications such as adrenal insufficiency (NR5A1, DAX1, StAR, CYP11A1, HSD3B2, and CYP17A1) and glomerulopathy or Wilms’ tumor (WT1).
Reproductive planning for the family
As with all genetic diseases, a molecular diagnosis informs risks of recurrence in other members of the family based on the mode of disease inheritance. This includes timely antenatal counseling and diagnosis, with pregnancy termination decisions backed by parents’ wishes and the severity of the disease in question.
MODALITIES OF GENETIC TESTING
In the following section, we review the various genetic tests that are available to guide diagnosis in a patient with DSD. While karyotype, FISH, and QF-PCR are used to delineate the type of DSD, the tests discussed subsequently are useful to pinpoint the exact molecular diagnosis in a patient with XY DSD. Table 2 provides a list of all investigations, their utilities, and their limitations.
| Modality | Utility | Resolution | TAT | Cost | Limitations |
|---|---|---|---|---|---|
| Karyotype | Chromosomal constitution | 5–10 Mb | 7–10 days | Rs 2,000 | Requirement of fresh sample, longer TAT, limited resolution |
| FISH | Chromosomal constitution and identification of CNVs | 50–500 Kb | 24–48 h | Rs 4,500 | Detects known chromosomal imbalances determined by specific probes, limited resolution |
| QF-PCR | Chromosomal constitution | Variable | Few hours | Rs 5,000 | Balanced rearrangements, partial monosomies, and low-level mosaicism cannot be detected; changes outside target sequences cannot be captured. |
| Sanger sequencing | Identification of SNVs in a highly specific phenotype, confirmation of NGS findings with suboptimal depth or coverage | 1 bp | 3–14 days | Rs 5,000 | Cumbersome sequence read is limited to 700-900 bp at a time and cannot identify variants in new genes |
| MLPA | Identification of CNVs in a highly specific phenotype, confirmation of CMA findings | 50–75 bp | 10-14 days | Rs 6,000 | Cannot identify balanced rearrangements, low-level mosaicism, and CNVs outside of the target region of interest |
| Chromosomal microarray | Identification of CNVs in gonadal dysgenesis and patients with a syndromic presentation | Up to 10 Kb | 14 days | Rs 10,000–15,000 | Cannot identify balanced chromosomal rearrangements and low-level mosaicism, challenging to interpret |
| WES | Identification of SNVs when the clinical phenotype is not suggestive, identification of novel DSD-related genes in protein coding regions | 1 bp | 4–6 weeks | Rs 10,000–15,000 | Cannot detect structural variation, sequencing depth may be insufficient for use in clinical settings |
| WGS | Provides a comprehensive picture of the entire genome, including SNVs, indels, CNVs, balanced translocations, inversions, aneuploidies | 1 bp | 10–12 weeks | Rs 60,000–80,000 | Expensive, extensive data storage space and bioinformatic expertise are required. Interpretation of variants in non-coding regions is challenging |
TAT: Turnaround time, FISH: Fluorescent in situ hybridization, QF-PCR: Quantitative-fluorescent polymerase chain reaction, MLPA: Multiplex ligation-dependent probe amplification, WES: Whole exome sequencing, WGS: Whole genome sequencing, CNVs: Copy number variants, SNVs: Single nucleotide variants
Karyotype
Karyotype is a conventional cytogenetic technique that provides a visual representation of chromosome complement after pre-treated chromosomes are subjected to condensation and Giemsa staining (G-banding). It provides information on alteration in chromosome number and structure provided that the changes are larger than 5,000–10,000 Kb in size. The technique requires that cells are cultured and arrested in metaphase, which is the most appropriate stage for visualizing and studying chromosomes in their most compact state. Guidelines recommend that a minimum of two metaphases must be thoroughly analyzed, although, in practice, many more cells are usually counted.[18] Due to the requirement of culture of fresh viable cells, the turnaround time ranges from 7 to 10 days.[19] Karyotype is usually the first genetic investigation that helps to classify DSD based on the underlying chromosomal constitution.
FISH
FISH is also a cytogenetic technique, but unlike karyotype, it has a much higher resolution of 50–500 Kb and a more rapid turnaround time of 24–48 h. It utilizes fluorescent probes directed against specific chromosomal regions to enable the identification of insertions, deletions, and translocations. Its utility in the context of DSD lies in the identification of the Y chromosome/SRY gene. Guidelines recommend the analysis of at least 50 interphase cells for reporting.[18,20] However, up to 600–2000 interphase cells can be analyzed in cases with a high index of suspicion of sex chromosomal DSD. Thus, FISH is a useful tool for quick identification of chromosomal sex (XX vs. XY), for detecting SRY gene translocation (as in 46, XX testicular or ovotesticular DSD), and for detection of mosaicism when the karyotype is 46,XX, or 45,X.
QF-PCR
QF-PCR is the most rapid method which relies on PCR-based amplification of sex chromosomes. The turnaround time is a few hours, and the results are available at the end of one working day. Among all modalities to identify sex chromosomes, QF-PCR is the fastest and most cost-effective. However, it has limited capabilities for the assessment of mosaicism and partial monosomies.[8,19]
Single gene testing
Testing of a single gene using Sanger sequencing is undertaken to pinpoint the exact molecular etiology. It is the preferred method when the patient’s phenotype and biochemical investigations are highly suggestive of an underlying genetic diagnosis, as is the case with steroid biosynthetic defects. Sanger sequencing becomes even more attractive if the concerned gene is either small in size or has known hotspots. For example, the SRD5A2 gene is small with 5 exons and has a well-described hotspot (p.R246Q) in the Indian subcontinent.[16,21] When deletion/duplication of a gene or its segment is a major disease mechanism, for example, in AIS, the test of choice is multiplex ligation-dependent-probe amplification (MLPA). MLPA is a kit-based test, with the kit being specific for particular genes, that is, AR in this case. The overall yield of single gene testing has been found to be poor (~10%), primarily due to significant phenotypic overlap among various disease entities.[22] However, it must be borne in mind that appropriate patient population selection is a major determinant of the diagnostic rate. For example, in a study by our group, 75 children with a high pre-test probability of 5α-reductase deficiency or PAIS were evaluated sequentially by Sanger sequencing of SRD5A2 and AR genes. Pathogenic/likely pathogenic variants in SRD5A2 were identified in 25/75 (33.3%) and in AR in 19/50 (38%).[16,23]
Testing for DSD panels
Over the past decade, advances in molecular techniques have led to the development of DSD panels comprising different numbers of DSD-related genes. Genes in such panels are sequenced using massive parallel high throughput sequencing techniques (e.g., NGS) to enable the identification of SNVs and small insertions and deletions (indels) in protein-coding regions of DSD-related genes. A diagnostic yield ranging from 31% to 46% has been obtained from studies utilizing DSD panels comprising 30–219 genes in patients with XY DSD.[22,24-29]
In different series, including ours, SRD5A2, AR, and NR5A1 have been the most frequently implicated genes in 46,XY DSD.[16,22,24-28,30,31]
CMA
CMA is used to identify microdeletions and microduplications well beyond the threshold of conventional cytogenomic techniques. At a resolution of 10 Kb, it can identify small intragenic as well as large structural CNVs. As opposed to MLPA, which identifies CNVs in a single gene, CMA scans the entire genome for deletions and duplications.[19,32] The two main types of CMA are single-nucleotide polymorphism array and array comparative genomic hybridization. The utility of this modality in the context of XY DSD lies mainly in the diagnosis of cases of gonadal dysgenesis, with an overall yield of ~30%; 5.6% in isolated gonadal dysgenesis, increasing all the way up to 25% in syndromic gonadal dysgenesis.[33] Etiologies such as DAX1 deletion (resulting in hypogonadotropic hypogonadism and adrenal hypoplasia congenita), DAX1 duplication (resulting in XY complete gonadal dysgenesis), and a deletion upstream of SOX9 in its regulatory region (resulting in XY gonadal dysgenesis) may be identified using CMA.[34] CMA has conventionally been an underutilized technique, with recent studies showing progressively greater yield [35,36] and recent guidelines placing it higher up in the diagnostic algorithm for the evaluation of DSD.[8,14,37-40]
WES
WES captures a nucleic acid sequence of the entire protein-coding region of the genome using high throughput sequencing technologies. The raw data are initially screened for relevant DSD genes and their immediately adjacent splice sites. In case no disease-causing variant is identified, the analysis proceeds to include other genes. Thus, WES has facilitated the discovery of novel genes and unraveled new phenotypes associated with previously reported genes (e.g., NR5A1).[34,40-43] WES has also brought into focus the phenomenon of oligogenic inheritance, wherein hits in multiple genes involved in the pathway to determine the final phenotype may culminate in the structural and functional manifestation in an individual. In XY DSD, oligogenic inheritance has been postulated for NR5A1, MAP3K1, AR, HSD3B2, WT1, WDR11, GATA4, DHH, etc.[44]
It is generally preferred to do WES in trios comprising the index patient and both parents. This increases the yield of testing as it allows several variants of uncertain significance (VUS) to be upgraded based on their presence or absence in symptomatic or asymptomatic parents.[45] However, this is not necessarily the case with DSDs, where the expression of many genes is sex-restricted.[28] For example, a boy with XY gonadal dysgenesis and a heterozygous variant in the NR5A1 gene may have inherited the variant from his asymptomatic mother, who may either continue to remain asymptomatic or go on to develop premature ovarian failure at a later age (not manifest at the time of testing). The major limitations of WES include the identification of VUS, detection of incidental findings unrelated to the phenotype that may pose significant ethical dilemmas, exclusion of non-coding regions, and inability to identify larger structural variants.
Whole genome sequencing (WGS)
Currently confined to the research setting, WGS is a powerful tool capable of sequencing the entirety of the human genome with accuracy and identifying both SNVs and CNVs in coding as well as non-coding regions of the genome. It is predicted to result in an impressive diagnostic yield for all genetic disorders. WGS comes with several limitations, though, the chief ones being its exorbitant cost, and enormous volume of data with increased cost of storage, increased complexity of analysis, and limited expertise in interpretation.[8]
Summary of genetic testing
Despite the armamentarium of currently available genetic tests, the choice of investigation is of utmost importance in obtaining a quick and accurate diagnosis and reducing the financial burden on patients’ families. The role of a focused clinical history and examination, appropriate baseline hormonal assays (especially when the patient presents in mini-puberty or puberty), and ultrasonography cannot be overemphasized. However, genetic testing can be given precedence over doing dynamic or expensive endocrine testing, and over imaging-based investigations that are invasive or require anesthesia.
When the suspicion of a monogenic etiology is extremely high, single gene testing using Sanger sequencing or MLPA may be undertaken depending on whether the gene is known to be primarily affected by SNVs or CNVs. In the absence of a clear diagnosis, multiple genes may be sequenced simultaneously (panel testing via NGS) or subject to CMA; with a lower threshold to opt for CMA in cases of multiple congenital malformations, developmental delay, and intellectual disability.[19] When a structured approach is adopted, the yield of genetic testing in XY DSD has been seen to improve significantly to 64%.[30] Ideally, before and after ordering any genetic test, and especially when ordering an exome or genome level test, in-depth pre- and post-test counseling sessions need to be organized with the clinical geneticist and the families, with provision of information on expectations from the genetic test; its cost, turnaround time, and fallacies; the need to verify the diagnosis with further testing such as parental segregation; the interpretation of the test result; and the usefulness of the test in predicting disease trajectory and in reproductive planning.[1,39,46]
Fallacies of genetic testing
The shortcomings of genetic testing in children with DSD can be broadly grouped under the following heads:
Limitations of current diagnostic capabilities
Routinely available genetic investigations cannot detect variants in non-coding regions of the genome and disruption of epigenetic regulation. The interpretation of VUS is another major challenge. Besides, there remain several undiscovered genes, and known genes with unknown roles in gonadal development. Continued research activity and collaboration will augment diagnostic yields in the future.
Limitations related to high variability in phenotypic expression
Genotype-phenotype correlation is lacking for many disease entities, such that a molecular diagnosis cannot always predict the future course. Despite the general understanding that children with XY DSD due to 5α reductase type 2 deficiency undergo pubertal virilization and must be reared as males, this is not always the case. Up to 70–90% of patients are raised as girls and the proportion that undergoes gender change to male ranges from 12–95% in different series.[47-49] Truncating homozygous variants with no residual enzyme activity in functional studies have been seen to be associated with variable clinical phenotypes ranging from the expected completely feminine external genitalia to just mild undervirilization. Furthermore, no difference has been observed in the degree of virilization in those who undergo gender change vis-à-vis those who continue to identify themselves as females.[50] We believe that one of the ways forward is to generate more phenotypic and natural history data based on molecular diagnoses from culturally diverse settings, to draw genotype-phenotype correlations, and identify other predictors of future adult gender identity.
CONCLUSION
In today’s era, molecular genetic testing has become crucial for ending diagnostic uncertainty and providing closure to patients and their families. A clinical geneticist can guide the choice of investigation and help with providing pre-and post-test counseling. Molecular testing does not replace but complements conventional baseline endocrinological and radiological investigations for a smooth, quick, and cost-effective diagnostic process for patients and their families. At present, there are several deficiencies in our knowledge of the natural causes of several molecular etiologies. The more we test, the more we will understand the diverse spectrum of phenotypes and trajectories of pubertal development, gender identity, etc., associated with different gene variants.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
Patient's consent not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91:554-63.
- [CrossRef] [Google Scholar]
- Epidemiology and initial management of ambiguous genitalia at birth in Germany. Horm Res. 2006;66:195-203.
- [CrossRef] [Google Scholar]
- Disorders of sex development in office practice. Indian J Pediatr. 2023;90:1030-7.
- [CrossRef] [Google Scholar]
- Overview of genetics of disorders of sexual development. Curr Opin Pediatr. 2015;27:675-84.
- [CrossRef] [Google Scholar]
- Disorders of sex development-novel regulators, impacts on fertility, and options for fertility preservation. Int J Mol Sci. 2020;21:2282.
- [CrossRef] [Google Scholar]
- Analysis of the Wilms' tumor suppressor gene (WT1) in patients 46,XY disorders of sex development. J Clin Endocrinol Metab. 2011;96:E1131-6.
- [CrossRef] [Google Scholar]
- WT1: A single gene associated with multiple and severe phenotypes. Endocr Metab Sci. 2023;13:100143.
- [CrossRef] [Google Scholar]
- Establishing a molecular genetic diagnosis in children with differences of sex development: A clinical approach. Horm Res Paediatr. 2023;96:128-43.
- [CrossRef] [Google Scholar]
- Classic and backdoor pathways of androgen biosynthesis in human sexual development. Ann Pediatr Endocrinol Metab. 2022;27:83-9.
- [CrossRef] [Google Scholar]
- Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2012;97:E257-67.
- [Google Scholar]
- P450 oxidoreductase deficiency: A systematic review and meta-analysis of genotypes, phenotypes, and their relationships. J Clin Endocrinol Metab. 2020;105:dgz255.
- [CrossRef] [Google Scholar]
- The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5:569-76.
- [CrossRef] [Google Scholar]
- The external genitalia score (EGS): A European multicenter validation study. J Clin Endocrinol Metab. 2020;105:dgz142.
- [CrossRef] [Google Scholar]
- The current state of diagnostic genetics for conditions affecting sex development. Clin Genet. 2017;91:157-62.
- [CrossRef] [Google Scholar]
- Serum AMH in physiology and pathology of male gonads. Int J Endocrinol. 2013;2013:128907.
- [CrossRef] [Google Scholar]
- Spectrum of pathogenic variants in SRD5A2 in Indian children with 46,XY disorders of sex development and clinically suspected steroid 5α-reductase 2 deficiency. Sex Dev. 2019;13:228-39.
- [CrossRef] [Google Scholar]
- UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development. Clin Endocrinol (Oxf). 2011;75:12-26.
- [CrossRef] [Google Scholar]
- European guidelines for constitutional cytogenomic analysis. Eur J Hum Genet. 2019;27:1-16.
- [CrossRef] [Google Scholar]
- The use of genetics for reaching a diagnosis in XY DSD. Sex Dev. 2022;16:207-24.
- [CrossRef] [Google Scholar]
- Section E9 of the American College of Medical Genetics technical standards and guidelines: Fluorescence in situ hybridization. Genet Med. 2011;13:667-75.
- [CrossRef] [Google Scholar]
- Phenotype, genotype and gender identity in a large cohort of patients from India with 5α-reductase 2 deficiency. Andrology. 2015;3:1132-9.
- [CrossRef] [Google Scholar]
- Diagnostic application of targeted next-generation sequencing of 80 genes associated with disorders of sexual development. Sci Rep. 2017;7:44536.
- [CrossRef] [Google Scholar]
- Clinical, biochemical, and molecular characterization of indian children with clinically suspected androgen insensitivity syndrome. Sex Dev. 2022;16:34-45.
- [CrossRef] [Google Scholar]
- Targeted next-generation sequencing identification of mutations in patients with disorders of sex development. BMC Med Genet. 2016;17:23.
- [CrossRef] [Google Scholar]
- Diagnostic yield of targeted gene panel sequencing to identify the genetic etiology of disorders of sex development. Mol Cell Endocrinol. 2017;444:19-25.
- [CrossRef] [Google Scholar]
- Rapid molecular genetic diagnosis with next-generation sequencing in 46,XY disorders of sex development cases: Efficiency and cost assessment. Horm Res Paediatr. 2017;87:81-7.
- [CrossRef] [Google Scholar]
- A large cohort of disorders of sex development and their genetic characteristics: 6 novel mutations in known genes. Eur J Med Genet. 2021;64:104154.
- [CrossRef] [Google Scholar]
- Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016;17:243.
- [CrossRef] [Google Scholar]
- Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD) Endocr Connect. 2019;8:100-10.
- [CrossRef] [Google Scholar]
- High molecular diagnosis rate in undermasculinized males with differences in sex development using a stepwise approach. Endocrinology. 2020;161:bqz015.
- [CrossRef] [Google Scholar]
- Exome sequencing for the diagnosis of 46,XY disorders of sex development. J Clin Endocrinol Metab. 2015;100:E333-44.
- [CrossRef] [Google Scholar]
- Genomic testing for diagnosis of genetic disorders in children: Chromosomal microarray and next-generation sequencing. Indian Pediatr. 2020;57:549-54.
- [CrossRef] [Google Scholar]
- Array-CGH analysis in patients with syndromic and nonsyndromic XY gonadal dysgenesis: Evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum Reprod. 2010;25:2637-46.
- [CrossRef] [Google Scholar]
- New technologies to uncover the molecular basis of disorders of sex development. Mol Cell Endocrinol. 2018;468:60-9.
- [CrossRef] [Google Scholar]
- Cytogenomic investigation of syndromic Brazilian patients with differences of sexual development. Diagnostics (Basel). 2023;13:2235.
- [CrossRef] [Google Scholar]
- Molecular and cytogenetic analysis of romanian patients with differences in sex development. Diagnostics (Basel). 2021;11:2107.
- [CrossRef] [Google Scholar]
- Under-reported aspects of diagnosis and treatment addressed in the Dutch-Flemish guideline for comprehensive diagnostics in disorders/differences of sex development. J Med Genet. 2020;57:581-9.
- [CrossRef] [Google Scholar]
- New genomic technologies: An aid for diagnosis of disorders of sex development. Horm Metab Res. 2015;47:312-20.
- [CrossRef] [Google Scholar]
- Society for Endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015) Clin Endocrinol (Oxf). 2016;84:771-88.
- [CrossRef] [Google Scholar]
- Genetics in endocrinology: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): Position paper of EU COST action BM 1303 'DSDnet' Eur J Endocrinol. 2018;179:R197-206.
- [CrossRef] [Google Scholar]
- Mutations involving the SRY-related gene SOX8 are associated with a spectrum of human reproductive anomalies. Hum Mol Genet. 2018;27:1228-40.
- [CrossRef] [Google Scholar]
- ZNRF3 functions in mammalian sex determination by inhibiting canonical WNT signaling. Proc Natl Acad Sci U S A. 2018;115:5474-9.
- [CrossRef] [Google Scholar]
- Pathogenic variants in the DEAHbox RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet Med. 2020;22:150-9.
- [CrossRef] [Google Scholar]
- Oligogenic origin of differences of sex development in humans. Int J Mol Sci. 2020;21:1809.
- [CrossRef] [Google Scholar]
- Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880-7.
- [CrossRef] [Google Scholar]
- Caring for individuals with a difference of sex development (DSD): A consensus statement. Nat Rev Endocrinol. 2018;14:415-29.
- [CrossRef] [Google Scholar]
- Genetic analysis of 25 patients with 5α-reductase deficiency in Chinese population. Biomed Res Int. 2020;2020:1789514.
- [CrossRef] [Google Scholar]
- Phenotypical, biological, and molecular heterogeneity of 5α-reductase deficiency: An extensive international experience of 55 patients. J Clin Endocrinol Metab. 2011;96:296-307.
- [CrossRef] [Google Scholar]
- Steroid 5α-reductase 2 deficiency. J Steroid Biochem Mol Biol. 2016;163:206-11.
- [CrossRef] [Google Scholar]
- Integrative and analytical review of the 5-alpha-reductase type 2 deficiency worldwide. Appl Clin Genet. 2020;13:83-96.
- [CrossRef] [Google Scholar]




