
Translate this page into:
A child with hypocalcemia – Looking for the bedside clues!

*Corresponding author: Nithya Thuruthiyath, Department of Pediatrics, Jubilee Mission Medical College and Research Institute, Thrissur, Kerala, India. nithshyam13@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Thuruthiyath N, Kumar TM. A child with hypocalcemia – Looking for the bedside clues! J Pediatr Endocrinol Diabetes 2022;2:90-1.
Abstract
A 4-year 2-month-old girl presented with painful spasms in both hands and feet. She was noticed to have excessive weight gain and mild developmental delay and was diagnosed with hypothyroidism during infancy. She had a stocky build and round facies, brachydactyly, and subcutaneous calcification. Laboratory investigations showed low serum calcium, high serum phosphorus, and elevated intact PTH levels. A provisional diagnosis of pseudohypoparathyroidism was made.
Keywords
Hypocalcemia
Pseudohypoparathyroidism
Subcutaneous calcification

INTRODUCTION
Parathyroid hormone deficiency or resistance is an important cause of symptomatic hypocalcemia in children. We report the case profile and relevant clinical images of a child with pseudohypoparathyroidism.The specific diagnosis should not be delayed due to lack of recognition of clinical characteristics and associated features of the disease.
CASE PROFILE
A 4-year 2-month-old girl, born as a second child of third-degree consanguineous marriage, presented with painful spasms in both hands and feet for 3 days. There was no history of seizures, hyperventilation, vomiting, fever, or drug intake. She was born at term with a birthweight of 2.8 kg and her neonatal period was uneventful. At 11 months of age, she was noticed to have excessive weight gain and mild developmental delay. On evaluation, she was diagnosed with hypothyroidism and was started on thyroxine supplements.
On examination, she had a stocky build and round facies with no dysmorphism [Figure 1a]. Her height was less than third centile and her weight was at the 15th centile. Her head size was normal. She had short stubby fingers [Figure 1b]. There was a hyperpigmented rough plaque with a wrinkled surface on the back of the left knee, which was suggestive of a subcutaneous calcification [Figure 1c and d]. The Chvostek and trousseau signs were negative. There was no oral candidiasis. Gross motor delay was noticed. System examination was unremarkable.
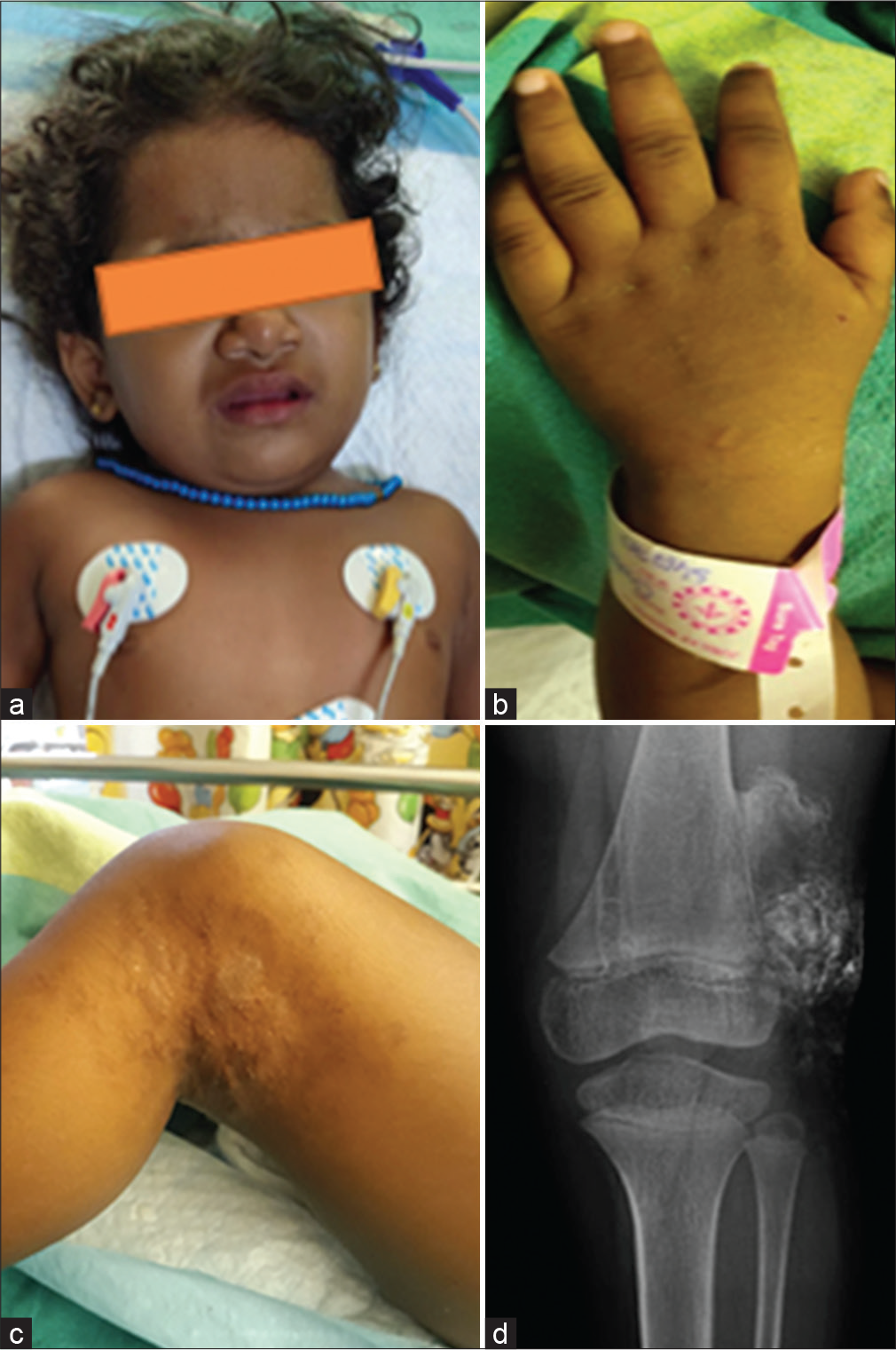
- (a) Clinical photograph of the child showing a stocky build with round facies. (b) Short stubby fingers with brachydactyly; no metacarpal sign was noted. (c) Hyperpigmented rough plaque with a wrinkled surface on the back of the left knee, suggestive of subcutaneous calcification. (d) X-ray of left knee confirming subcutaneous calcification.
Laboratory investigations showed serum calcium 5.80 mg/dL, phosphorus 9.30 mg/dL, magnesium 2.0 mg/dL, and alkaline phosphatase 141 IU/L. Liver function tests were normal with normal serum albumin levels. Renal function tests were normal. Venous blood gas studies showed normal pH. Thyroid evaluation showed TSH 3.48 µIU/mL and FT4 0.8 ng/dL. Serum vitamin D level was 25 ng/mL. Intact PTH assay done was high, 484 pg/mL. Urine calcium-tocreatinine ratio was normal (0.02).
A provisional diagnosis of pseudohypoparathyroidism was made. Considering the physical characteristics of the Albright hereditary osteodystrophy (AHO) phenotype and associated central hypothyroidism, pseudohypoparathyroidism (PHP) type 1a was considered.
Intravenous calcium gluconate correction was given under cardiac monitoring, along with vitamin D (800 IU/day) and calcitriol (0.07 µg/kg/day). Her serum calcium level improved and she was symptomatically better. She was sent home on oral calcium, calcitriol, and vitamin D. Genetic study could not be done. She was doing well at the first follow-up visit and had no spasms or muscle cramps. The last serum calcium level was 9.2 mg/dL with serum phosphorus 6 mg/dL.
DISCUSSION
PHP is a rare inherited disorder characterized by target organ resistance or unresponsiveness to parathyroid hormone.[1]
PHP is classified as either type 1 or type 2. Type 1 is characterized by the abnormal cAMP response to G protein activation, whereas the cAMP response is normal in type 2.[2] Type 1 is further subdivided into 1a, 1b, or 1c. PHP 1a (the most common subtype) and 1c exhibit multihormone resistance, and both express a phenotype of AHO. Children with AHO phenotype present with short stature, frequently below normal intelligence, obesity, round face, short neck, subcutaneous ossifications, brachydactyly, and shortened metatarsals.[1] PHP 1b is localized only to the kidney. Thus, the presence of the AHO phenotype or subcutaneous calcification during clinical evaluation should prompt a biochemical workup to confirm and differentiate the type of PHP.
CONCLUSION
Pseudohypoparathyroidism is a rare clinically heterogenous group of disorder with multihormone resistance. A careful clinical examination and laboratory work-up will be helpful in timely diagnosis and treatment of this condition.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Diagnosis and management of pseudohypoparathyroidism and related disorders: First International Consensus Statement. Nat Rev Endocrinol. 2018;14:476-500.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular definition of pseudohypoparathyroidism variants. J Clin Endocrinol Metab. 2021;106:1541-52.
- [CrossRef] [PubMed] [Google Scholar]




