
Translate this page into:
Dent disease type 2 due to OCRL1 mutation in a boy with rickets


*Corresponding author: Ganesh Jevalikar, Department of Endocrinology and Diabetes, Max Healthcare, New Delhi, India. gjevalikar@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Jevalikar G, Rana AS, Mithal A, Sethi SK. Dent disease type 2 due to OCRL1 mutation in a boy with rickets. J Pediatr Endocrinol Diabetes. doi: 10.25259/JPED_46_2024
Abstract
We report a case of an Indian boy who presented with rickets and was thought to have nutritional vitamin D deficiency rickets precipitated by the use of anticonvulsant medications. The presence of proteinuria and lack of normalization of alkaline phosphatase prompted further evaluation which was suggestive of type 2 Dent disease secondary to mutations in the OCRL1 gene. The case highlights the importance of urine routine, a simple yet useful test in the evaluation of rickets.
Keywords
Dent disease
OCRL1
Rickets

INTRODUCTION
Nutritional vitamin D deficiency is the most common cause of rickets worldwide. However, genetic forms of rickets are common, the most common being X-linked hypophosphatemic rickets (XLHR).
Dent disease is an X-linked recessive tubulopathy characterized by low-molecular-weight proteinuria, hypercalciuria, and nephrocalcinosis most commonly caused by mutations in CLCN5 gene (about 65% of cases), known as Dent disease type 1 (DD-1). In about 10–15% of cases, this can result from OCRL1 mutations known as Dent disease type 2 (DD-2). In other cases, genetic etiology is unknown.[1]
We report here a case of a boy who presented with rickets and was diagnosed with DD-2 secondary to OCRL1 mutation. To the best of our knowledge, this has not been reported from India previously.
CASE REPORT
A 12 ½-year-old Indian boy presented with a history of slow height gain for 4 years and progressive knee deformity with pain and difficulty in walking for 6 months. He had an episode of complex partial seizure 10 months before his presentation and was documented to have multiple ring-enhancing lesions on the neuroimaging in the right frontotemporal cortical and subcortical region. This was managed with anti-tubercular medications, glucocorticoids, and carbamazepine. Three months after the initiation of these medications, he was noted to have progressive genu valgum. There was no history suggestive of malabsorption, hepatic disorder, polyuria, or renal disease. He consumed 500 mL of milk daily and had limited sunlight exposure in recent months. For his complaints, he was treated with 120,000 IU of cholecalciferol orally without any biochemical testing. There was family history of similar bone deformities in a maternal male cousin. However, no clinical or laboratory evaluation was done. There was no history of consanguinity. Perinatal history was uneventful and developmental milestones were achieved at appropriate age.
On examination, his height was 124.6 cm (−3.53 standard deviation [SD]) and he weighed 35.7 kg (−0.7 SD). Blood pressure was normal. He had severe genu valgum [Figure 1a] with an inter-malleolar distance of 18 cm. Except for mild wrist widening, other bones were unremarkable. Significant dental caries were observed. Ophthalmic evaluation was negative for cataract. Systemic examination was unremarkable.
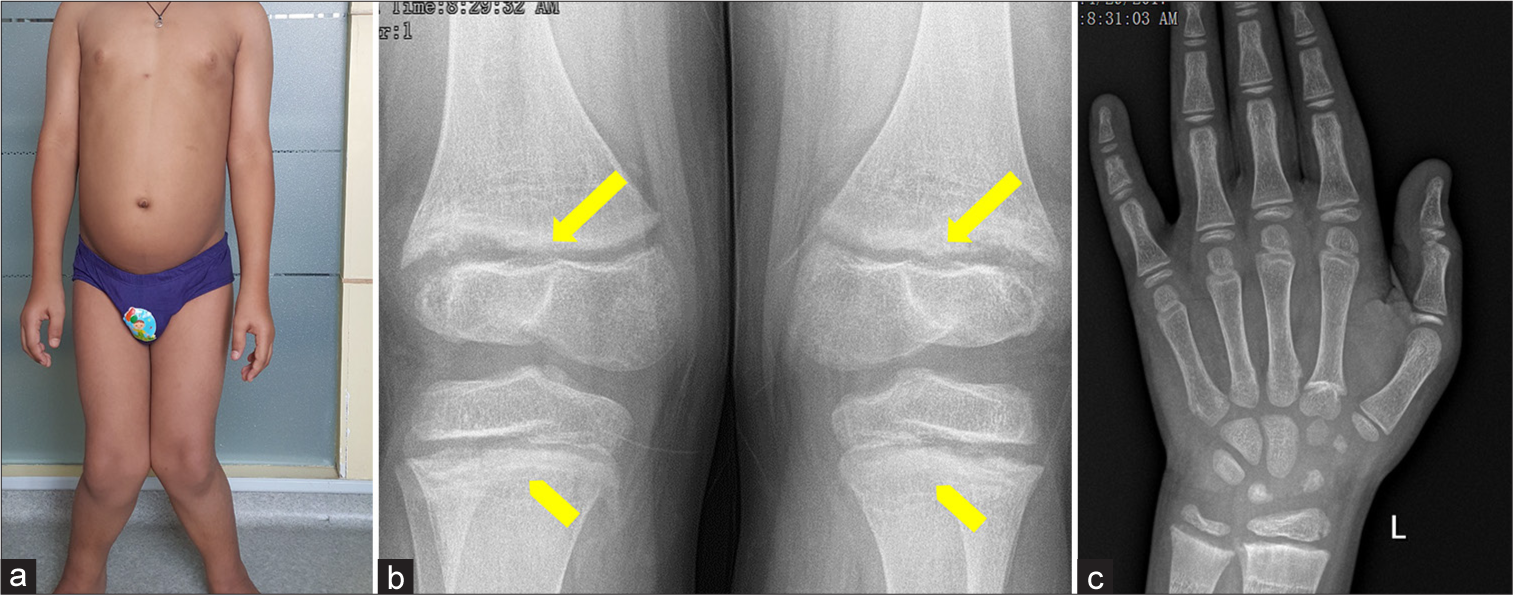
- (a) Severe genu valgum, along with metaphyseal flaring (yellow block arrows) and metaphyseal lucencies (yellow chevron arrows) in the (b) knee and (c) hand radiograph.
A differential diagnosis of drug-induced rickets versus other causes of non-nutritional rickets including renal tubular acidosis and hypophosphatemic rickets were considered. Radiographs of the knee and hand demonstrated mild fraying [block arrows, Figure 1b] and metaphyseal lucencies [Figure 1b and 1c]. Biochemistry [Table 1] revealed elevated serum alkaline phosphatase (ALP), low normal calcium and phosphorus, normal 25-hydroxyvitamin D (25-OHD), secondary hyperparathyroidism, and appropriately elevated 1,25-dihydroxyvitamin D [1,25(OH)2D] levels. Venous blood gas, liver, and kidney function tests were normal, except for mild proteinuria.
| Parameter | Normal range | Baseline | 3 months | 12 months |
|---|---|---|---|---|
| Height (cm) | 124.6 | 127.8 | 133.6 | |
| Weight (kg) | 35.7 | 37 | 40.9 | |
| Testes | 8 mL each | 15 mL each | ||
| Ca (mg/dL) | 8.8–10.7 | 8.9 | 9.6 | 10.1 |
| Phosphorus | 3.5 | 5.9 | 5.1 | |
| ALP (U/L) | 129–417 | 1402 | 910 | 674 |
| 25-OHD (ng/mL) | 20–100 | 32.6 | 35 | |
| PTH (pg/mL) | 10–65 | 197.7 | 47.5 | 30.3 |
| Urine Calcium/Creatinine | <0.2 | 0.25 | - | |
| 1,25(OH)2D (pmol/L) | 47–190 | 312 | - | |
| Treatment | D3 2,000 IU/day Calcium 1 g/day | D3 60,000 IU once a month Calcium 500 mg/day | D3 60,000 IU once a month Calcium 500 mg/day |
ALP: Serum alkaline phosphatase, PTH: Parathyroid hormone, 25-OHD: 25-hydroxyvitamin D, 1,25(OH)2D: 1,25-dihydroxyvitamin D3, D3 - Cholecalciferol
Since the 25-OHD levels were documented after cholecalciferol and there was a distinct history of knee deformity developing after the medications received for ring-enhancing lesions, a diagnosis of partially treated vitamin D and calcium deficiency was considered, and it was decided to continue vitamin D along with calcium [Table 1]. With this treatment, there was a distinct clinical and biochemical improvement in the form of the disappearance of knee pains, an increase in height of 9 cm over 1 year, normalization of calcium, phosphorus, parathyroid hormone (PTH) levels, and a decline (but not complete normalization) in the levels of ALP. In view of the lack of complete normalization of ALP and significant short stature, further evaluation was performed.
A repeat urinalysis showed persistent proteinuria (urine protein 278 mg/dL, urine protein/creatinine ratio of 3.43) with normal serum albumin level (5 g/dL) despite normal PTH. On further analysis, β2-microglobinuria (32977 ng/mL, normal range <300) along with hypercalciuria (236 mg/kg/day, normal range <4 mg/kg/day) was documented. Renal ultrasound was negative for nephrocalcinosis. Clinical exome sequencing followed by Sanger sequencing confirmed a novel hemizygous 5’ splice site pathogenic variation in intron 7 of the OCRL gene (chrX: 128692731G>T; Depth: 478x) that affects the invariant GT donor splice site of exon 7 (c.560+1G>T; ENST00000371113). The same variant was also found in the mother. This confirmed the diagnosis of DD-2 in this patient. This mutation was not previously described in 1000 genomes and Exome Aggregation Consortium databases. Based on in silico prediction, the variant was classified as a pathogenic mutation.
He was further managed with calcium and vitamin D supplementation in maintenance doses, with monitoring to rule out worsening of hypercalciuria along with guided growth surgery with bilateral medial distal femur hemiepiphysiodesis with eight plates for the deformities.
DISCUSSION
This is the first reported case of OCRL1 mutation causing Dent disease from India. The mutation found in our case has not been reported previously. Hypophosphatemia often thought to be the cause of rickets in Dent disease was absent in our case. The case also highlights thinking beyond nutritional rickets with a lack of complete response to vitamin D and calcium supplements and the role of urine routine, a simple yet useful investigation in the evaluation of rickets.
Nutritional vitamin D and/or calcium deficiency is the most common cause of rickets worldwide. Among hereditary causes of rickets, XLHR is one of the most common.[2] In the present case, initially, rickets was thought to be nutritional rickets possibly aggravated by anticonvulsants. Prior treatment with a mega-dose of cholecalciferol made the interpretation of serum 25-OHD levels difficult. Hence, treatment was continued with calcium and maintenance doses of vitamin D. Although there was a good clinical response to the treatment given, the serum ALP levels did not normalize even after a year. The presence of severe short stature with a lack of normalization of the ALP levels prompted the evaluation of alternative causes of rickets in this case.
The presence of proteinuria and family history of maternal male cousin with similar disorder raised a possibility of X-linked renal tubular disorder. Since XLHR does not have proteinuria, Dent disease was considered in the index child and confirmed by further biochemical testing which confirmed low-molecular-weight proteinuria (LMWP) and hypercalciuria. Genetic testing confirmed the diagnosis of DD-2. The case also highlights the importance of urine routine as a simple but useful test in the evaluation of children with suspected non-nutritional rickets.
Dent disease is an X-linked disorder with variable degrees of proximal renal tubular dysfunction characterized by LMWP, hypercalciuria, and nephrocalcinosis. Nearly 60% of cases are due to mutations in CLCN5 gene (DD-1) whereas 15–20% of cases are due to mutations in OCRL1 gene (DD-2). In other cases, genetic etiology is unknown.[3] There is no specific treatment for Dent disease. Thiazide diuretics are often used effectively to reduce hypercalciuria but may be accompanied with complications of hypovolemia and hyponatremia. Angiotensin-converting enzyme (ACE) inhibitors/angiotensin receptor blockers are effective in reducing proteinuria although there is no evidence of their benefit in the reduction of progression to end-stage renal disease which sets in usually after the second decade of life.
Rickets has been described in about 20% cases of Dent disease.[4] However, this percentage is likely to be much higher in India. In an Indian cohort of 18 patients, all patients had evidence of rickets. Among 15 patients who had genetic testing, 13 had CLCN5 mutations but none had OCRL1 mutation.[5] To the best of our knowledge, this is the first reported case of genetically proven OCRL1 mutation causing DD-2 from India. Hypophosphatemia was absent in our case which is consistent with the fact that it is present only in 10% cases of DD-2.[1] Treatment of rickets with calcium and vitamin D supplements has to be done with careful monitoring to prevent the worsening of hypercalciuria.
Mutations in OCRL1 gene cause oculocerebrorenal syndrome of Lowe as well as DD-2. There are phenotypic differences in these two conditions in the form of presence of renal tubular acidosis, hypotonia, severe developmental delay, and congenital cataracts in the former.[3] A genotypephenotype correlation is suggested with mutations in exons 1–7 being associated with DD-2 and those with truncating mutations in exons 8–24 with Lowe syndrome.[6] A recent case series of Fanconi renotubular syndrome from India reported one patient with OCRL1 mutation who had Lowe syndrome. Rickets was not seen in this patient probably due to hypotonia and non-weight bearing.[7] The mutation of this patient was in exon 16, whereas in our patient, the mutation was located in exon 7, further supporting the genotypephenotype correlation.
It may not be possible to differentiate DD-1 from DD-2 on the clinical basis as the renal phenotype overlaps. Although a study from Japan reported higher height SD scores, higher prevalence of chronic kidney disease (defined as Cr-eGFR <90 mL/min/1.73 m2), higher levels of creatinine kinase, lactate dehydrogenase, and aspartate transaminase in DD-2 compared to DD-1.[8] Glycosuria, hypophosphatemia, hypokalemia, hypercalciuria, and nephrocalcinosis are more frequently described in DD-1. Failure to thrive is more common in DD1 whereas intellectual disability is more common in DD-2.[1]
CONCLUSION
A broad differential diagnosis needs to be considered in all cases of rickets in addition to nutritional rickets and XLHR. Family history, short stature, proteinuria, and lack of normalization of biochemical parameters should alert the clinician to non-nutritional rickets. Urinalysis can be a simple but useful investigation to be done in all cases of suspected non-nutritional rickets.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Genetics and phenotypic heterogeneity of Dent disease: The dark side of the moon. Hum Genet. 2021;140:401-21.
- [CrossRef] [PubMed] [Google Scholar]
- Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260-7.
- [CrossRef] [PubMed] [Google Scholar]
- Observations of a large Dent disease cohort. Kidney Int. 2016;90:430-9.
- [CrossRef] [PubMed] [Google Scholar]
- Phenotype of dent disease in a cohort of Indian children. Indian Pediatr. 2016;53:977-82.
- [CrossRef] [PubMed] [Google Scholar]
- OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol. 2009;112:27-36.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited Fanconi renotubular syndromes: Unveiling the intricacies of hypophosphatemic rickets/osteomalacia. J Bone Miner Metab. 2024;42:155-65.
- [CrossRef] [PubMed] [Google Scholar]
- Comparison of clinical and genetic characteristics between Dent disease 1 and Dent disease 2. Pediatr Nephrol. 2020;35:2319-26.
- [CrossRef] [PubMed] [Google Scholar]




