
Translate this page into:
Molecular mechanisms underlying congenital hyperinsulinemia of infancy and its relevance to management – A review

*Corresponding author: Seema Kapoor, Department of Pediatrics, Maulana Azad Medical College and Associated Lok Nayak Hospital, Delhi, India. drseemakapoor@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mittal M, Gupta AK, Kapoor S. Molecular mechanisms underlying congenital hyperinsulinemia of infancy and its relevance to management – A review. J Pediatr Endocrinol Diabetes. 2024;4:9-20. doi: 10.25259/JPED_25_2024
Abstract
Congenital hyperinsulinemia of infancy (CHI), characterized by inappropriate insulin secretion despite low blood glucose, is by far the most common cause of persistent hypoglycemia in infancy. The presentation is typically in the first few days of life and could be life-threatening. A critical sample drawn at the time of hypoglycemia is crucial for biochemical characterization and is the beginning of a cascade of investigations that further elucidate our course of action. The majority of the cases relate to defects in KATP channels that regulate insulin secretion from pancreatic beta-cells. These are mostly attributable to mutations in ABCC8 and KCNJ11, both located on the short arm of chromosome 11, that code subunits of the KATP channel (sulfonylurea receptor [SUR] and Kir6.2, respectively). However, the underlying molecular defect may be identified in only about half of them. Much before the molecular diagnosis is established, therapy needs to be initiated. Diazoxide is the initial choice as it acts on the KATP channels at SUR1 and opens them, preventing insulin release. The involvement of the pancreas may be diffuse or focal. The diffuse form arises from dominant or recessive mutations affecting the KATP channel. The recessive ones are more common and cause the more severe forms of CHI. Where diazoxide proves ineffective, other interventions, such as octreotide, may be tried. If hypoglycemia remains unresolved despite all medical therapy, a near-total pancreatectomy would be required. On the other hand, focal involvement of a specific group of beta-cells results from paternally inherited germinal mutation together with post-zygotic loss of normal maternal allele. Elective partial pancreatectomy in these focal cases would completely ameliorate hypoglycemia. Hence, based on the genotype, one can plan further diagnostic modalities such as fluorine 18L-3,4 dihydroxyphenylalanine positron emission tomography scan to define whether the involvement is diffuse or focal and consider the management accordingly.
Keywords
Hypoglycemia
diazoxide
K-ATP channel
ABCC8
KCNJ11

INTRODUCTION
Glucose is the essential metabolic fuel for the fetal brain, for which it is dependent entirely on the mother. With the clamping of the cord at birth, this supply is interrupted, the glucose levels drop, insulin secretion reduces, and that of counter-regulatory hormones increases. The glucose levels drop in the initial few hours to stabilize by 72 hours of life.[1] The newborn brain, being proportionately large, consumes a larger amount of the energy supply and is at risk of neuronal injury even with transient hypoglycemia.[1]
CASE SCENARIOS
Case 1
A 3.5 kg female baby born of third-degree consanguineous marriage had repeated episodes of seizures since day 1 of life. Blood glucose (BG) was documented to be <45 mg/dL at these episodes. She received a continuous infusion of dextrose-containing fluids but continued to have hypoglycemic episodes with seizures. The glucose infusion rate was increased to 12 mg/kg/min. A “critical” sample taken at the time of hypoglycemia revealed pH 7.3, bicarbonate 23 mEq/mL, blood ketones tested by ketostix 0.5 mmol/L, and no reducing substances in the urine. Administration of glucagon 0.1 mg IV caused a brisk rise of BG by 45 mg/dL. Insulin was detected at 14.4 µIU/mL, and C-peptide was 1 ng/mL. A diagnosis of hyperinsulinism was made, and diazoxide was started at a dose of 10 mg/kg/day and gradually increased to 20 mg/kg/day. However, the baby still needed a high glucose infusion rate to maintain normoglycemia. Considering diazoxide-unresponsiveness, an injection of octreotide was started as an infusion on which the BG improved, and the baby shifted to subcutaneous injections. Molecular genetic analysis by Sanger sequencing revealed a homozygous deletion of exon 7 in the ABCC8 gene. The parents were heterozygous for this variation. The parents were explained the inheritance of the condition and counseled for the chances of its reoccurrence in future conceptions. Unfortunately, the parents could not continue with a further hospital stay, and later, we learned that the baby died soon after.
Case 2
A 3.6 kg male baby, born of non-consanguineous marriage, had repeated episodes of hypoglycemia since the 1st day of life. He presented with seizures and BG was 35 mg/dL at that time. He required high rates of infusion of glucose at more than 10 mg/kg/min to maintain normoglycemia. A critical sample showed BG 40 mg/dL, insulin 2.78 µIU/mL, cortisol 17.10 ng/mL, ketones 0.2 mmol/L, no acidosis, and brisk response to IV glucagon. With a presumptive diagnosis of hyperinsulinemia, diazoxide was initiated, but hypoglycemia did not resolve even at 20 mg/kg/day. Intravenous infusion of octreotide was started and increased to 18 µg/kg/day to maintain normoglycemia. Genetic analysis revealed a heterozygous mutation in ABCC8 with uniparental disomy. ABCC8 splicing variant c.4415–13G>A was detected on one allele, and the second ABCC8 variant was not found. The mutation was inherited from the father. A focal lesion was very likely, and a fluorine 18L-3,4 dihydroxyphenylalanine positron emission tomography (18F-DOPA PET) scan was performed. and it revealed a focal lesion at the junction of the body and tail of the pancreas [Figure 1]. A focal pancreatectomy was performed, and the hypoglycemia resolved completely.
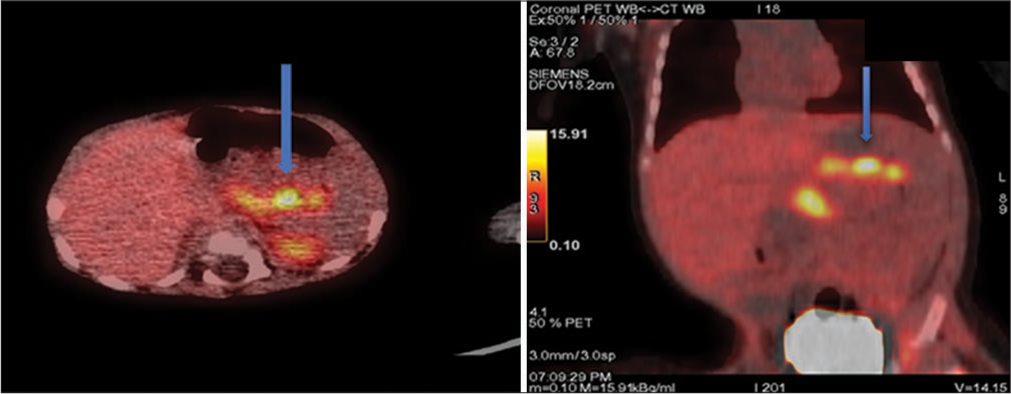
- Fluorine 18L-3,4 dihydroxyphenylalanine positron emission tomography scan images of pancreas showing increased radiotracer uptake (shown by arrows) at junction of body and tail of pancreas suggesting focal lesion. (Case and image credit: Dr Rajni Sharma, Additional Professor, AIIMS, New Delhi).
What are the major forms of neonatal hypoglycemia?
The three major forms of neonatal hypoglycemia are as follows:
Transitional hypoglycemia – This affects all neonates in the first few days of life. The low glucose threshold for insulin secretion that occurs in fetal life persists into postnatal life, resulting in transitional hypoglycemia.
Perinatal stress-induced hypoglycemia in high-risk neonates – The perinatal stress causes an exaggeration of the low glucose threshold for insulin release, resulting in hyperinsulinemia
Hyperinsulinemia due to genetic defects – This refers to increased insulin release due to mutations that affect the insulin release pathway, the majority acting through inactivation of the ATP-sensitive potassium (KATP) channels.
It is important to recognize that hyperinsulinemia is common to all these major causes of hypoglycemia.[1] At the same time, other significant causes of hypoglycemia, such as fatty acid oxidation defects, hypopituitarism, and glycogen storage disorder, must be ruled out.
CONGENITAL HYPERINSULINEMIA OF INFANCY (CHI)
What is the incidence of CHI, and how is it classified?
The incidence of CHI is estimated at 1:50,000 live births, though it is much higher in populations with consanguineous marriages.[2] It can be classified as:
Channelopathies – These involve defects in ion channels located on the beta(β)-cell membrane, commonly affecting the KATP channels. Mutations affecting the voltage-gated calcium channels (VGCAs) also present with hyperinsulinemia
Metabolopathies – These involve defects in the metabolic pathways leading to insulin release. These include activating mutations (e.g., for glutamate dehydrogenase [GDH]) or inactivating mutations (as for hydroxyacyl coenzyme A dehydrogenase) of enzymes that affect insulin release.
What is the molecular mechanism of insulin release?
The KATP channel is composed of four subunits of sulfonylurea receptor 1 (SUR1) and four subunits of Kir6.2 (inward rectifier potassium channel).[3] The Kir6.2 forms the pore of the channel, while SUR1 subunits act as regulatory subunits. The genes coding for the KATP channel include ABCC8, which codes for SUR1, and KCNJ11, which codes for Kir6.2, both localized to chromosome 11p15.1.[4,5]
The secretion of insulin from the β-cells is directly affected by the BG concentrations. With elevated BG concentrations, the glucose uptake by β-cells increases, and the rate of glucose oxidation increases, resulting in an increased ATP/ADP ratio. This inhibits the SUR1 subunits, causing the closure of KATP channels. The cell membrane gets depolarized, and VGCA opens, causing an influx of Ca2+ that causes exocytosis of insulin-laden storage granules [Figure 2]. On the other hand, when glucose concentrations are low, the KATP channels open, allowing K+ to diffuse freely, the membrane potential remains hyperpolarized, and no insulin release occurs.[6]
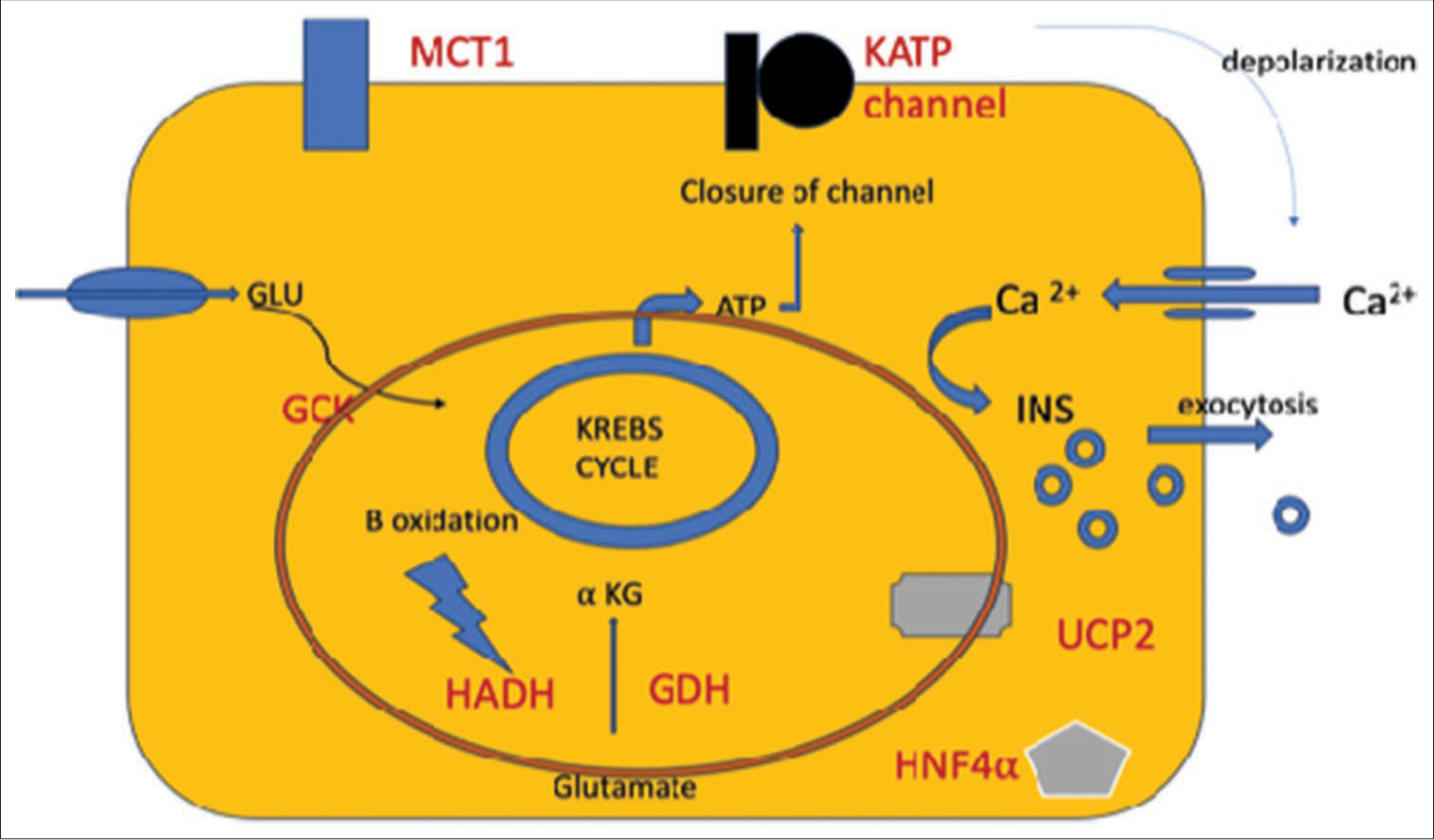
- Diagrammatic representation of molecular mechanisms causing CHI in a pancreatic beta-cell. (The red oval represents the mitochondria). CHI: Congenital hyperinsulinemia of infancy, αKG: Alpha-ketoglutarate, ATP: Adenosine triphosphate, GCK: Glucokinase, GDH: Glutamate dehydrogenase, GLU: Glucose, HADH: Hydroxyacyl-coenzyme A dehydrogenase, HNF4α: Hepatocyte Nuclear Factor 4-alpha, INS: Insulin, KATP channel: ATP-sensitive potassium channel, MCT1: Monocarboxylate transporter 1, UCP2: Uncoupling protein 2.
Why is there a risk of neurologic injury in hyperinsulinemia?
Insulin increases the intracellular uptake of glucose, inhibits gluconeogenesis and glycolysis, and inhibits lipolysis and ketone body production. With hypoglycemia and the absence of alternative energy sources (ketones), the brain is at high risk of hypoglycemic brain injury. However, there is no correlation between the level of serum insulin and the severity of hypoglycemia.[7]
What are the clinical features of hyperinsulinemic hypoglycemia?
These neonates are born macrosomic (as a result of the growth-promoting effect of insulin), and symptoms start early in life, within a few days of life, or sometimes later in infancy. The symptoms include lethargy, poor feeding, apnea, hypothermia, seizures, and altered sensorium. They may have hepatomegaly and cardiomegaly as a result of fetal hyperinsulinemia and storage of glycogen.[2,8] The requirement of high glucose infusion rates beyond 8 mg/kg/min is almost synonymous with CHI.
What is the pathologic classification of CHI?
CHI could be classified into three types on the basis of β-cell involvement:
Diffuse – The β-cells that hypersecrete insulin is present throughout the pancreas. This accounts for 60–70% of all cases.
Focal – There is nodular hyperplasia of islet-like cell clusters, including dutculoinsular complexes and giant nuclei at a localized area of the pancreas, with surrounding pancreatic tissue being normal. This accounts for 30–40% of cases
Atypical – This could be described as a combination of the above two patterns.
The identification of the histological type may be crucial, as the focal type responds well to focal pancreatectomy with preservation of the exocrine pancreas and the uninvolved endocrine part as well. The gold standard test is the 18F-DOPA PET scan. It has a sensitivity of 89% and a specificity of 98% in differentiating the diffuse and focal forms.[2]
What is the genotype-phenotype correlation seen in KATP CHI?
CHI can be categorized into familial and non-familial forms. Monogenic forms of familial CHI are primarily attributed to genetic defects in the pathways regulating insulin secretion from pancreatic β-cells. Many genes have been known to play a significant role in familial monogenic CHI, including ABCC8, KCNJ11, GLUD1, glucokinase (GCK), hydroxyacylcoenzyme A dehydrogenase (HADH), uncoupling protein 2 (UCP2), INSR, HNF1A, HNF4A, SLC16A1, and PGM1. ADK, ALG3, AKG6, CACNAIC, CACNA 1D, CDKN1C, CREBBP, DIS3L2, EIF253, EP300, FAH, Forkhead box protein (FOXA2), GPC3, HK1, HRAS, KCNJ11, KCNQ1, KMT2D, KDM6A, and MPI.[9,10]
Recessive inactivating mutations in ABCC8 and KCNJ11 are the most common causes of CHI and result in reduced activity or complete loss of function of the KATP channels. Persistent unregulated insulin release despite low BG causes severe hypoglycemia. More than 150 homozygous, compound heterozygous, and heterozygous inactivating mutations in ABCC8 and more than 24 in KCNJ11 have been reported.[11] These cause defects in KATP channel synthesis, turnover, or transport from the endoplasmic reticulum and Golgi apparatus to the plasma membrane or alteration in open-state frequency. They typically cause diffuse CHI, need high glucose infusion rates, and are unresponsive to diazoxide. Dominant inactivating mutations usually present a milder phenotype and respond to diazoxide, though they may sometimes be more severe and medically unresponsive.[6]
The focal form involves two independent events. The first is the inheritance of a paternal heterozygous mutation in ABCC8 or KCNJ11, followed by the second event, which is the bodily loss of the maternal 11p allele (11p15.1–11p15.5), with consequent loss of cell repressor genes, forming the focal lesion. This paternal uniparental disomy unmasks the paternally inherited KATP channel mutation, which leads to altered expression of a number of imprinted genes, including the maternally expressed tumor suppressor genes H19 and CDKN1C and the paternally expressed growth factor insulin-like growth factor 2.[12,13] These result in focal proliferation of ß-cells, that lack the KATP channel, leading to focal adenomatous hyperplasia.[14] The focal involvement is always sporadic in origin.[6,12]
Atypical CHI is characterized by morphological mosaicism with hyperfunctional islets restricted to certain pancreatic areas, making it difficult to classify as a diffuse or localized type.[15,16] While one of the genetic pathways for chromosome 11p15.1 with an ABCC8 gene mutation has been shown to be explained by mosaic interstitial paternal uniparental isodisomy,[15] the genetic basis of the majority of individuals with atypical CHI is still unknown.[16]
What are the other molecular mechanisms causing CHI?
GDH
GDH is a mitochondrial enzyme that catalyzes the oxidative deamination of glutamate to alpha-ketoglutarate and ammonia. Alpha-ketoglutarate enters the Krebs cycle, and the generation of ATP triggers further molecular mechanisms leading to exocytosis of insulin. An increased activity of this enzyme as a result of gain of function mutation in GLUD1 leads to increased insulin release and hyperinsulinemia with hypoglycemia [Table 1]. The mutation is dominant and de novo in origin. It results in reduced sensitivity of GDH to inhibition by GTP and ATP. Since leucine can activate GDH, it may precipitate after protein-rich meals. The birth weight is normal, and the hypoglycemia presents in late infancy or childhood and is usually mild and provoked by fasting or high-protein meals. As GDH presents in the liver and also gets unduly active, there is increased production of ammonia and reduced urea synthesis. Hyperammonemia is usually asymptomatic and does not need treatment[17]. Known as hyperinsulinism-hyperammonemia syndrome, it is the second most common cause of CHI after KATP channel defects. There is diffuse involvement of the pancreas. As GDH lies upstream of the KATP pathway, diazoxide is usually effective. There is a high likelihood of the child developing developmental delay, learning disabilities, and epilepsy.
| Disorder | Gene |
|---|---|
| Monogenic forms | |
| KATP channel defect | ABCC8, KCNJ11 |
| GDH–HI/HA | GLUD1 |
| GCK | GCK |
| HADH | HADH1 |
| UCP2 | UCP2 |
| HNF4α | HNF4A |
| HNF1α | HNF1A |
| Pyruvate transporter – MCT1 | SLC16A1 |
| HK1 | HK1 |
| PGM | PGM1 |
| Syndromic forms | |
| Beckwith–Wiedemann syndrome | IGF2, H19, CDKN1C, and KCNQ1 |
| Sotos syndrome | NSD1 |
| Usher syndrome | Contiguous gene deletion affecting ABCC8 |
| Congenital disorders of glycosylation (GDG 1a, 1b, 1d) | PMM2, MPI, and ALG3 |
| Simpson–Golabi–Behmel syndrome | GPC3 and GPC4 |
| Perlman syndrome | DIA3L2 |
| Timothy syndrome | CACNA1C; |
| Insulin resistance syndrome | INS |
| Congenital central hypoventilation syndrome | PHOX2B |
| Growth failure syndromes | |
| Kabuki syndrome | KMT2D and KDM6A |
| Costello syndrome | HRAS |
| Chromosomal disorders | |
| Mosaic turner syndrome | Loss of X in some cells |
| Patau syndrome | Trisomy 13 |
GCK: Glucokinase, GDH: Glutamate dehydrogenase, HADH: Hydroxyacyl-coenzyme A dehydrogenase, HI/HA: Hyperinsulinism-hyperammonemia syndrome, HK: Hexokinase, HNF1α: Hepatocyte Nuclear Factor 1-alpha, HNF4α: Hepatocyte nuclear factor 4-alpha, MCT1: Monocarboxylate transporter 1, PGM: Phosphoglucomutase, UCP2: Uncoupling protein 2, IGF: Insulin-like growth factor, PMM2: Phosphomannomutase 2, CHI: Congenital hyperinsulinemia of infancy
GCK
The GCK (or hexokinase [HK] IV) enzyme, coded by GCK, catalyzes the phosphorylation of glucose to form glucose-6-phosphate. This enters the glycolysis pathway, producing ATP that signals the closure of KATP channels in the pancreatic β-cells, and through membrane depolarization and the influx of calcium, insulin is released. The enzyme has a high affinity for glucose at relatively high glucose concentrations. Gain of function mutations, transmitted as autosomal dominant, lowers the threshold of glucose-mediated insulin release.
The first presentation is usually after the age of 6 months, though it could be anytime from birth to adulthood, as fasting hypoglycemia, with a family history of an affected parent.[18] The pancreatic involvement is diffuse. The response to diazoxide is variable, though the majority do respond.[19]
Variability in presentation can be observed within families with the same variant.
HADH
HADH is an intramitochondrial enzyme involved in the β-oxidation pathway. Decreased activity of this enzyme leads to increased activity of GDH, increased ATP production, and insulin release. Loss of function in HADH is recessively inherited and is the most common genetic defect causing CHI in consanguineous births.[20] The pancreatic involvement is diffuse. The severity is variable, ranging from severe neonatal hypoglycemia to milder later-onset hypoglycemia induced by protein intake and response to diazoxide.
Hepatocyte nuclear factor 4alpha (HNF4α)
This belongs to the hormone receptor superfamily, and inactivating mutations in the encoding gene may cause a reduction in the expression of KATP channel subunit Kir6.2 or a reduction in the levels of PPARα.[21,22] The latter effect causes a decrease in β-oxidation of fatty acids and accumulation of malonyl coenzyme A in the cytosol, which inhibits carnitinepalmitoyltransferase 1. This increases the concentration of long-chain acyl-CoA levels, signaling insulin release.[23] The phenotypic presentation is quite variable, ranging from transient neonatal CHI to persistent hypoglycemia, macrosomia, and often family history of maturity-onset diabetes in young. The histological involvement is diffuse and responds to diazoxide.
UCP2
Present on the inner mitochondrial membrane, this protein reduces the ATP/ADP ratio in β-cells. The KATP channel remains open, and insulin secretion is reduced. Loss of function in the encoding gene leads to hyperinsulinemia. Another possible mechanism is by modulating the production of reactive oxygen species.[24] The hypoglycemia is usually mild.
FOXA2
This transcription factor is required for organogenesis and development of both the pancreas and pituitary gland as they are both endoderm-derived. A mutation in FOXA2 leads to an imbalance in pancreatic β/α-cell ratio, profound hypoglucagonemia, and hyperinsulinemia. FOXA2 also regulates the expression of KCNJ11 and ABCC8 genes and has a role in the transactivation of the HADH gene.[25] It presents as hypopituitarism, CHI, and dysmorphism along with liver, pancreas, and heart and gastrointestinal abnormalities.
Calcium voltage-gated channel subunit alpha-1D (CACNA1D)
The CACNA1D gene encodes the VGCAs that are expressed in pancreatic β-cells, and the influx of calcium causes exocytosis of insulin. An activating mutation causes the channels to remain open at a lower membrane potential, leading to dysregulated insulin secretion. There are associated heart defects and severe hypotonia.[26]
HK1
Similar to the action of GCK, it phosphorylates glucose to form glucose 6-phosphate. It has a high affinity for glucose at low glucose levels. Its expression is silenced at low glucose levels to prevent insulin release. Dominant gain of function mutations has been reported to cause inappropriate insulin release.[27]
Monocarboxylate transporter 1
Encoded by the SLC16A1 gene mapped on chromosome 1p13.2-p12, it transports monocarboxylates (pyruvate and lactate) into the cells. Dominant activating mutations cause increased pyruvate uptake, increased ATP production, and, thus, increased insulin release.[28] The condition is characterized by hypoglycemic episodes, which usually occur within 30–45 min after exercise, in response to the accumulation of pyruvate and lactate. Hence, carbohydrate intake before and during exercise is important to prevent episodes of hypoglycemia. The defect is upstream of the KATP pathway, and diazoxide usually works.
What are the metabolic conditions leading to CHI?
Congenital disorders of glycosylation (CDG)
These are a group of multisystem disorders that involve inherited defects in the synthesis of glycan moiety of glycoconjugates or in the attachment of the glycan to macromolecules. The glycoconjugates have crucial roles in intracellular metabolism and protein structure and function. Although the precise mechanism leading to hyperinsulinemia has not been elucidated, abnormal glycosylation at the level of the sulfonylurea receptor may be the underlying mechanism. CDG should be considered a possible cause in cases of CHI without a clear etiology.
CDG Ia, the most common type, is caused by mutations in the phosphomannomutase 2 gene. Among its varied clinical features, hyperinsulinemia is not a frequent manifestation. CDG Ib, with phosphomannose-isomerase deficiency, presents with hyperinsulinemic hypoglycemia as a leading symptom with other features such as protein-losing enteropathy, congenital hepatic fibrosis, and coagulopathy without overt neurologic manifestations that are commonly seen in other CDGs.[29] Oral mannose supplementation alleviates symptoms, and hence, early diagnosis is crucial. CDG Id has also been described to present with HI. Most cases of CDGs do respond to diazoxide.[6]
Tyrosinemia type I and CHI
CHI may be part of the symptomatology of tyrosinemia type 1, though the precise mechanism is not known. This could be related to the accumulation of toxic metabolites due to a deficiency of the enzyme fumaryl acetoacetate hydrolase. Severe liver and kidney disease are the main features, and hypoglycemia persists despite dietary treatment.[30] Diazoxide does improve hypoglycemia.
What are the syndromes associated with CHI?
While CHI mostly presents as an isolated entity, it may be associated with certain syndromes, most commonly seen with Beckwith–Wiedemann syndrome. Typical features such as prenatal and postnatal onset macrosomia, macroglossia, organomegaly, and hemihypertrophy may be present. Early neonatal hypoglycemia is present in 50% of these cases with variable diazoxide responsiveness.[31]
Kabuki syndrome is characterized by various dysmorphic features, such as elongated palpebral fissures with eversion of the lateral lower eyelid. Thick finger pads may be associated with CHI in <7% of cases that usually respond to diazoxide. Other syndromes with hyperinsulinemia include Turner syndrome, Sotos syndrome, and others listed in Table 1. The exact mechanism causing hypoglycemia is not well understood. They generally respond to diazoxide and resolve with time.[19]
What is postprandial hyperinsulinemia (PPHI)?
PPHI typically occurs a few hours after meals in children who have undergone Nissen fundoplication/gastric bypass. These children have an exaggerated secretion of glucagon-like peptide 1 (GLP1), which acts as an insulin secretagogue, causing exaggerated insulin surge and hypoglycemia.[32] PPHI has also been reported in children with insulin autoimmune syndrome. Children newly exposed to exogenous insulin develop insulin-binding autoantibodies.
What is insulinoma?
Insulinoma, or insulin-secreting tumor of the pancreas, is a rare cause of hyperinsulinism in children or adolescents.[33] The intracellular mammalian target of the rapamycin (mTOR) pathway is involved in increased insulin secretion. Its possibility should be considered in children older than 2 years with acquired hyperinsulinemia and CT or MRI should be done. Careful family history is important as it could be part of multiple endocrine neoplasia. Surgical removal of the tumor is indicated.
What is the diagnostic approach to CHI?
Clinical evaluation
As detailed above, babies with CHI are macrosomic. The presence of dysmorphic features and organomegaly points toward syndromic forms. It is important to take a history of consanguinity and loss of siblings or a history of similar illness in siblings. Typical symptoms of hypoglycemia would be present as lethargy, poor feeding, or seizures. Short periods of fasting leading to severe hypoglycemia and the requirement of glucose infusion rate exceeding 8–10 mg/kg/min for maintaining normoglycemia are strong pointers toward hyperinsulinemia.
Laboratory evaluation
A critical sample drawn at the time of hypoglycemia is crucial to reaching the diagnosis [Box 1]. High levels of insulin are not always detectable, and the insulin/glucose ratio is not always reliable. Any measurable insulin at times of plasma glucose <50 mg/dL is considered suggestive of hyperinsulinemia, as it would otherwise be undetectable. Absence of ketosis, acidosis, free fatty acids, and elevated C-peptide is other suggestive evidence. Administration of glucagon (30 µg/kg IV or IM, maximum 1 mg) induces a brisk rise (within an hour) in plasma glucose (measured bedside every 15 min) more than 30 mg/dL from baseline and is considered diagnostic (in the absence of multiple pituitary hormone deficiencies).[10] This differentiates hyperinsulinemia from other conditions, such as glycogen storage disease and gluconeogenic and glycogenolytic defects, where the rise would be <30 mg/dL. If a particular stimulus is suspected to trigger hypoglycemia, the tests may be carried out at the time of hypoglycemia after protein loading or exercise, as the case may be.
Biochemical tests
Hormones and metabolites
Urine
|
Further evaluation
Once the diagnosis of CHI is reached, diazoxide is initiated as it is a KATP channel opener. The dose is 5–15 mg/kg/day in three divided doses. If doses of 15 mg/kg/day or more for 5 days do not resolve the hypoglycemia, it is considered diazoxide-unresponsive. Other medical therapies are initiated, and genetic testing is ordered to look for mutations typical of focal CHI, in which case radionuclide imaging with 18F-DOPA PET/CT is performed [Figure 3]. It is based on the principle of selective uptake and conversion into dopamine by the DOPA decarboxylase enzyme, which is present in β-cells. There is sustained retention of the radiotracer by hyperplastic clonally expanded β-cells as compared to the rest of the pancreas.
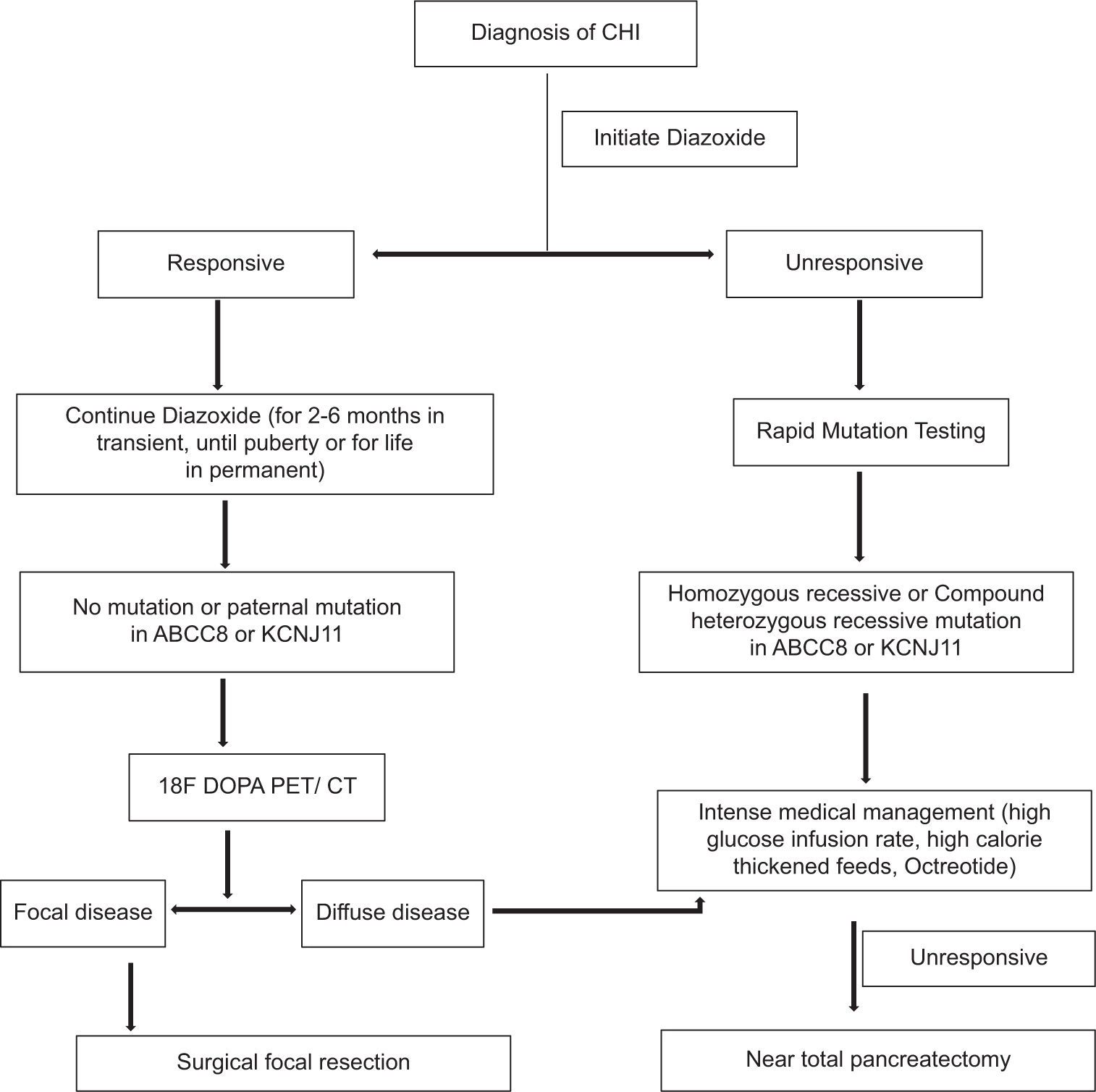
- Diagnostic and management plan for congenital hyperinsulinemia of infancy.
What is the approach to genetic testing in CHI?
Genetic testing is of paramount importance for making molecular diagnosis of CHI and the recent guidelines recommend it for all patients with hyperinsulinemia persisting beyond 3 months age (except those likely to have an acquired cause).[34] However, it is typically unwarranted for clinical management in diazoxide-responsive patients, especially in resource constrained settings. The yield of genetic testing in CHI patients ranges from 45.3% to 79%, and the detection rate of diazoxide-unresponsive CHI patients is almost 90%.[34,35] In cases where CHI is associated with a histologically diffused form, next-generation sequencing (NGS) is typically the initial test indicated, followed by Sanger validation.[36,37]
NGS with copy number variation (CNV) analysis is increasingly being employed for the screening of small copy number variants, providing an accessible and cost-effective option. In instances where a mutation remains undetected in blood samples, and pancreatectomy has been conducted, re-evaluating the known CHI genes to identify variants exclusive to pancreatic DNA warrants consideration.
Methylation studies should be considered for cases exhibiting histologically focal variants or syndromic variants like Beckwith-Wiedemann syndrome. Karyotyping remains the gold standard genetic test for conditions such as CHI associated with monosomy X, trisomy 13, and other chromosomal disorders suspected in certain cases.
Multiplex-ligation-dependent probe amplification (MLPA) has limited utility in the identification of deletions and duplications in individual genes, for example, ABCC8, KCNJ11, GLUD1, GCK, HADH, UCP2, INSR, HNF1A, HNF4A, and SLC16A1. MLPA has enormous significance in detecting mosaicism, particularly in conditions where low-level somatic mosaicism plays a role in disease manifestation or progression.
Microarray-based comparative genomic hybridization (array CGH) is very useful for the identification of large deletions, duplications, and aneuploidies; in contrast to MLPA, array CGH lacks the capability to detect low-level mosaicism, particularly mosaicism below 30% for deletions and duplications, and below 10% for aneuploidies. Nevertheless, this method permits the examination of CNV across the whole genome.
Beyond offering insights into coding and non-coding regions, genome sequencing enables the detection of structural changes in copy number variants, including large deletions, duplications, aneuploidies, and mosaic variants. Nonetheless, the sensitivity of this approach for detecting low-level mosaic variants is diminished compared to targeted NGS, attributed to the lower read depth achieved. Long-read sequencing holds promise for concurrently detecting sequence variation and DNA methylation status, although their clinical utilization remains limited. It has primarily been employed for genes challenging to sequence using conventional methodologies.
Optical genome mapping, as a next-generation cytogenetic technique, holds incredible potential. However, its widespread clinical use requires further validation.
Can genetic testing guide the dietary management?
GLUD1 gene-associated hyperinsulinism-hyperammonemia syndrome may benefit from a protein-restricted diet to manage both hypoglycemia and hyperammonemia. Similarly, mutations in the HADH gene, which cause hyperinsulinism due to short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency, may respond to a diet high in carbohydrates and low in fat, as well as frequent feeding to prevent hypoglycemia.[38]
In cases where genetic testing reveals mutations that are associated with diazoxide-unresponsive CHI, such as those in the ABCC8 or KCNJ11 genes, dietary management in conjunction with medical therapy or surgical intervention is of immense importance. Somatic mutations within the GCK gene represent a rare yet significant etiology of diazoxide-unresponsive atypical CHI.[39]
What is the role of genetic counseling?
It provides families with information about the nature of the disorder, the implications for the affected individual, and the risks for other family members. This is essential for making informed decisions about family planning and management options. If a child has a recessive form of CHI, there is a 25% chance of recurrence in each subsequent pregnancy if both parents are carriers. In cases of dominant mutations, the risk of recurrence can be 50% if one parent is affected. However, if the mutation is de novo, the risk of recurrence is significantly lower but not zero due to the possibility of germline mosaicism.
Paternally inherited missense variations in the ABCC8 or KCNJ11 genes can lead to a severe form of CHI with an early presentation.
What is the management of CHI?
Acute management
The neonate/infant would need dextrose bolus followed by continuous glucose infusion at rates >8 mg/kg/min to maintain normoglycemia. Glucagon 1 mg bolus or infusion could be life-saving, acutely raising BG level, as it mobilizes glycogen stores from the liver. Other supportive management would be necessary, including establishing secure central venous access. Frequent/continuous feedings need to be given, which often necessitates a gastrostomy.
Long-term management
Medical management is tried first, followed by surgical options. The first drug of choice is diazoxide, which requires a functional KATP channel to bind onto. It is ineffective in recessive mutations that cause the channel to be inactivated, as in diffuse CHI, as well as in most cases of focal CHI. Since it is not possible to clinically differentiate between the two entities, an 18F-DOPA PET scan is necessary, as detailed above. Other forms of hyperinsulinemia, such as due to risk factors such as intrauterine growth restriction and perinatal asphyxia, respond to diazoxide, as do most syndromic cases. It has fluid retaining properties and could cause hyponatremia, tachypnea, congestive heart failure with pulmonary edema and pulmonary hypertension. Hence thiazide diuretics, like chlorothiazide, that also have synergistic action over the channels are added. Long term side effects include hypertrichosis and coarsening of facial features. Other rare side effects include neutropenia, thrombocytopenia and hyperuricemia. Response to diazoxide should be evaluated after at least 5 days as it has a long half-life. Responsiveness would imply a withdrawal of IV glucose infusion and normoglycemia on oral feeds, fasting capability adequate for age, suppressed insulin, and appropriate levels of ketones at the end of the fast. Screening echocardiography should be done before initiating treatment and 1 week after that. Blood counts and uric acid levels should be tested every 6 months.
Feeds should be supplemented with glucose to increase carbohydrate content to 15%. Addition of uncooked corn starch to feeds provides stability to glucose control in infants and older children.
In diazoxide-unresponsive cases, nifedipine may be tried until surgery can be scheduled [Table 2]. Although there are some reports of success in reaching normoglycemia and are considered an add-on drug, larger studies do not encourage its use. It acts by blocking the calcium channels in the beta-cells.[40]
Octreotide may be added to diazoxide therapy in partially responsive cases or as a second-line agent in cases of unresponsiveness. It causes hyperpolarization of β-cells and also inhibits calcium channels. Prolonged use causes the internalization of receptors and reduces the effectiveness of the drug.[41] A long-acting analog, lanreotide, has good efficacy.
For cases of PPHI, frequent feeds containing long-acting carbohydrates (corn starch), abundant protein, fiber, and fat need to be administered, and if still there is hypoglycemia, acarbose that slows the absorption of glucose into the bloodstream is tried.[42] It avoids the glycemic peak that is followed by exaggerated insulin release in these patients.
It has been hypothesized that mTOR complex 1 may be overactivated in diffuse CHI. Inhibitors of the mTOR pathway have been used to treat insulinoma. The use of sirolimus (previously known as rapamycin) in CHI was first reported by Senniappan et al., and there are numerous case reports of its use in CHI after that.[43,44] Sirolimus has shown good efficacy in CHI due to ABCC8 and KCNJ11, although Al-Balwi et al. have reported reduced effects in those with homozygous mutations.[45] There are reports where surgery could be delayed for 6–12 months.[46,47] There are concerns over its immunosuppressive effect that leads to frequent infections and hepatotoxicity. It could also cause pancreatic insufficiency and diabetes mellitus.[47]
Novel therapies
GLP-1 antagonists (exendin 9–39) have been tried in adults with CHI due to KATP mutations with considerable improvement and have potential for future use in younger age groups. It opposes the insulin secretagogue action of GLP-1.[48]
| Drug | Route | Dose | Side effects |
|---|---|---|---|
| Diazoxide | Oral | 5–20 mg/kg/day | Sodium and water retention |
| Hypertrichosis | |||
| Loss of appetite | |||
| Chlorothiazide | Oral | 5–10 mg/kg/day (divided in 2 doses) | Hyponatremia |
| Hypokalemia | |||
| Glucagon | Bolus: SC/IM | Bolus: 0.02 mg/kg/dose | Skin rash |
| Infusion: SC/IV | Infusion:5–10 µg/kg/h | Vomiting | |
| Doses beyond recommended range may stimulate insulin release and cause rebound hypoglycemia | |||
| Nifedipine | Oral | 0.25–2.5 mg/kg/day (divided in 2–3 doses) | Hypotension |
| Octreotide | Infusion: SC/IV | 5–35 µg/kg/day (divided in 3–4 doses or continuous infusion) | Vomiting |
| Raised liver enzymes | |||
| Long QT syndrome | |||
| Necrotizing enterocolitis | |||
| Sirolimus | Oral | Starting dose: 0.5 mg/m2/day (divided in 2 doses) | Frequent infections |
| Adjust dose aiming for blood concentrations 5–15 ng/mL | Raised liver enzymes Hyperlipidemia |
CHI: Congenital hyperinsulinemia of infancy, SC: Subcutaneous, IM: Intramuscular, IV: Intravenous
Surgical management
Surgical removal of the focal lesion in focal CHI achieves a complete cure in most cases. Intraoperative biopsies are taken to ensure that the excision margins are histologically clear. A laparoscopic approach may be feasible for accessible areas (body or tail of the pancreas), while open laparotomy would be required for those in the head of the pancreas.[49,50]
For diffuse CHI unresponsive to medical management, a near-total pancreatectomy would be required. The resolution of hypoglycemia is, however, not without potential future complications of diabetes mellitus and exocrine pancreatic insufficiency.[51]
Follow-up
It is important to keep these children under regular follow-up to monitor their neurodevelopmental outcome, apart from monitoring their compliance to treatment and development of any side effects. Deficits are known to develop in domains of speech and language, motor, and vision. Some may develop seizures, including infantile spasms.[52]
CONCLUSION
CHI is a significant condition that needs to be recognized and diagnosed urgently. A comprehensive workup is essential, and timely management can save lives and prevent sequelae as well. While the initial drug of choice is diazoxide, it is important to be aware that most of the mutations would not respond to it, and alternates need to be started. Imaging to look for focal hyperplasia needs to be considered so that a surgical approach can be planned.
Acknowledgments
The authors would like to thank Dr. P. S. N. Menon, former Professor of Pediatrics at All India Institute of Medical Sciences (AIIMS), New Delhi, and Consultant and Head of the Department of Pediatrics, Jaber Al-Ahmed Armed Forces Hospital, Kuwait, for his valuable inputs in the preparation of the manuscript and to Dr. Rajni Sharma, Additional Professor Pediatrics, Division of Pediatric Endocrinology, AIIMS, New Delhi for contributing to the manuscript preparation and contributing the case scenario 2.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
Patient consent was not required as there are no patients in this study.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- New approaches to screening and management of neonatal hypoglycemia based on improved understanding of the molecular mechanism of hypoglycemia. Front Pediatr. 2023;11:1071206.
- [CrossRef] [PubMed] [Google Scholar]
- Reconstitution of KATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995;270:1166-70.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital hyperinsulinaemic hypoglycaemia-a review and case presentation. J Clin Med. 2022;11:6020.
- [CrossRef] [PubMed] [Google Scholar]
- Nesidioblastosis no longer! It's all about genetics. J Clin Endocrinol Metab. 2011;96:617-9.
- [CrossRef] [PubMed] [Google Scholar]
- Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. J Clin Endocrinol Metab. 2016;101:815-26.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinaemic hypoglycaemia: Genetic mechanisms, diagnosis and management. J Inherit Metab Dis. 2012;35:589-601.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinism in infancy and childhood: When an insulin level is not always enough. Clin Chem. 2008;54:256-63.
- [CrossRef] [PubMed] [Google Scholar]
- Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000;82:F98-107.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital hyperinsulinism in infancy and childhood: Challenges, unmet needs and the perspective of patients and families. Orphanet J Rare Dis. 2022;17:61. Erratum in: Orphanet J Rare Dis 2022;17:205
- [CrossRef] [PubMed] [Google Scholar]
- International guidelines for the diagnosis and management of hyperinsulinism. Horm Res Paediatr. 2024;97:279-98.
- [CrossRef] [PubMed] [Google Scholar]
- Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat. 2009;30:170-80.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013;98:E355-63.
- [CrossRef] [PubMed] [Google Scholar]
- Unbalanced expression of 11p15 imprinted genes in focal forms of congenital hyperinsulin-ism: Association with a reduction to homozygosity of a mutation in ABCC8 or KCNJ11. Am J Pathol. 2001;158:2177-184.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital hypoglycemia disorders: New aspects of etiology, diagnosis, treatment and outcomes: Highlights of the proceedings of the congenital hypoglycemia disorders symposium, Philadelphia, April 2016. Pediatr Diabetes. 2017;18:3-9.
- [CrossRef] [PubMed] [Google Scholar]
- An ABCC8 gene mutation and mosaic uniparental isodisomy resulting in atypical diffuse congenital hyperinsulinism. Diabetes. 2008;57:259-63.
- [CrossRef] [PubMed] [Google Scholar]
- Morphological mosaicism of the pancreatic islets: A novel anatomopathological form of persistent hyperinsulinemic hypoglycemia of infancy. J Clin Endocrinol Metab. 2011;96:3785-93.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinism-hyperammonaemia syndrome: Novel mutations in the GLUD1 gene and genotypephenotype correlations. Eur J Endocrinol. 2009;161:731-5.
- [CrossRef] [PubMed] [Google Scholar]
- Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58:1419-27.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinemic hypoglycemia in children and adolescents: Recent advances in understanding of pathophysiology and management. Rev Endocr Metab Disord. 2020;21:577-97.
- [CrossRef] [PubMed] [Google Scholar]
- Genome-wide homozygosity analysis reveals HADH mutations as a common cause of diazoxide-responsive hyperinsulinemic-hypoglycemia in consanguineous pedigrees. J Clin Endocrinol Metab. 2011;96:E498-502.
- [CrossRef] [PubMed] [Google Scholar]
- The MODY1 gene HNF-4alpha regulates selected genes involved in insulin secretion. J Clin Invest. 2005;115:1006-15.
- [CrossRef] [PubMed] [Google Scholar]
- Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology. 2005;146:375-82.
- [CrossRef] [PubMed] [Google Scholar]
- Malonyl-CoA signalling, lipid partitioning, and glucolipotoxicity: Role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51(Suppl 3):S405-13.
- [CrossRef] [PubMed] [Google Scholar]
- Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: Mechanism of action. Diabetes. 2001;50:1302-10.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital hyperinsulinism and hypopituitarism attributable to a novel mutation in FOXA2. J Clin Endocrinol Metab. 2018;103:1042-7.
- [CrossRef] [PubMed] [Google Scholar]
- A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes. 2017;18:320-3.
- [CrossRef] [PubMed] [Google Scholar]
- Dominant form of congenital hyperinsulinism maps to HK1 region on 10q. Horm Res Paediatr. 2013;80:18-27.
- [CrossRef] [PubMed] [Google Scholar]
- The genetic and molecular mechanisms of congenital hyperinsulinism. Front Endocrinol (Lausanne). 2019;10:111.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinemic hypoglycemia-leading symptom in a patient with congenital disorders of glycosylation 1a (phosphomannomutase deficiency) J Inherit Metab Dis. 2001;24:858-62.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinism in tyrosinaemia type I. J Inherit Metab Dis. 2005;28:131-5.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinism and BeckwithWiedemann syndrome. Arch Dis Child Fetal Neonatal Ed. 2001;84:F67-9.
- [CrossRef] [PubMed] [Google Scholar]
- Increased glucagon-like peptide-1 secretion and postprandial hypoglycemia in children after Nissen fundoplication. J Clin Endocrinol Metab. 2009;94:39-44.
- [CrossRef] [PubMed] [Google Scholar]
- International guidelines for the diagnosis and management of hyperinsulinism. Horm Res Paediatr. 2024;97:279-98.
- [CrossRef] [PubMed] [Google Scholar]
- Navigating the U.S. health insurance landscape for children with rare diseases: A qualitative study of parents' experiences. Orphanet J Rare Dis. 2021;16:313.
- [CrossRef] [PubMed] [Google Scholar]
- The CHI centers of excellence program. 2021. Available from: https://congenitalhi.org/wp-content/uploads/2021/07/coe-press-release.pdf [Last accessed on 2024 Jun 07]
- [Google Scholar]
- Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376-80.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the noncoding genome. Curr Opin Pediatr. 2015;27:659-64.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital hyperinsulinemia of infancy: Role of molecular testing in management and genetic counseling. Indian J Pediatr. 2022;89:395-8.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlation in Taiwanese children with diazoxide-unresponsive congenital hyperinsulinism. Front Endocrinol. 2023;14:1283907.
- [CrossRef] [PubMed] [Google Scholar]
- Assessment of nifedipine therapy in hyperinsulinemic hypoglycemia due to mutations in the ABCC8 gene. J Clin Endocrinol Metab. 2017;102:822-30.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term medical treatment in congenital hyperinsulinism: A descriptive analysis in a large cohort of patients from different clinical centers. Orphanet J Rare Dis. 2015;10:150.
- [CrossRef] [PubMed] [Google Scholar]
- Pharmacokinetic-pharmacodynamic relationships of Acarbose. Clin Pharmacokinet. 1996;30:94-106.
- [CrossRef] [PubMed] [Google Scholar]
- Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N Engl J Med. 2014;370:1131-7.
- [CrossRef] [PubMed] [Google Scholar]
- Sirolimus therapy in congenital hyperinsulinism: A successful experience beyond infancy. Pediatrics. 2015;136:e1373-6.
- [CrossRef] [PubMed] [Google Scholar]
- Sirolimus in the treatment of three infants with diffuse congenital hyperinsulinism. J Pediatr Endocrinol Metab. 2017;30:1013-7.
- [CrossRef] [PubMed] [Google Scholar]
- Hyperinsulinemic hypoglycemia in infancy: Current concepts in diagnosis and management. Indian Pediatr. 2015;52:1051-9.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical efficacy evaluation of sirolimus in congenital hyperinsulinism. Int J Endocrinol. 2020;2020:7250406.
- [CrossRef] [PubMed] [Google Scholar]
- GLP-1 receptor antagonist exendin-(9-39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes. 2012;61:2585-91.
- [CrossRef] [PubMed] [Google Scholar]
- Laparoscopic vs open pancreatectomy for persistent hyperinsulinemic hypoglycemia of infancy. J Pediatr Surg. 2009;44:957-61.
- [CrossRef] [PubMed] [Google Scholar]
- Pancreatic head resection and Roux-en-Y pancreaticojejunostomy for the treatment of the focal form of congenital hyperinsulinism. J Pediatr Surg. 2012;47:130-5.
- [CrossRef] [PubMed] [Google Scholar]
- Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care. 2012;35:198-203.
- [CrossRef] [PubMed] [Google Scholar]
- Abnormal neurodevelopmental outcomes are common in children with transient congenital hyperinsulinism. Front Endocrinol (Lausanne). 2013;4:60.
- [CrossRef] [PubMed] [Google Scholar]




