
Translate this page into:
Late bloomer: Delayed diagnosis of Prader-Willi syndrome

*Corresponding author: Dhanya Soodhana, Department of Pediatric and Adolescent Endocrinology, Aster Malabar Institute of Medical Sciences, Kozhikode, Kerala, India. dhanyasoodhana@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Marakkar S, Soodhana D. Late bloomer: Delayed diagnosis of Prader-Willi syndrome. J Pediatr Endocrinol Diabetes. 2024;4:42-4. doi: 10.25259/JPED_19_2024
Abstract
Prader–Willi syndrome (PWS) commonly presents with hypotonia and feeding difficulties in infancy or rapid weight gain during childhood. We present a girl with PWS who was diagnosed for the 1st time when she presented with primary amenorrhea.
Keywords
Delayed puberty
Prader–Willi syndrome
Hypogonadism
Obesity

INTRODUCTION
Prader Willi Syndrome (PWS) represents the most common form of syndromic obesity. Disorders of pubertal development are common in PWS and depending on the age they could present with genital hypoplasia, delayed puberty or incomplete gonadal maturation.[1] The diagnosis should not be delayed due to lack of recognition of clinical characteristics.
CASE DESCRIPTION
A 16-year-old girl presented with primary amenorrhea despite having breast development and pubic hair growth since the age of 11 years. Her mother observed excess weight gain starting at the age of 10 years, along with short stature compared to peers, disturbed sleep, and daytime sleepiness. There were no reported headaches, vomiting, convulsions, trauma, galactorrhea, or prolonged drug intake. She is the first-born child of non-consanguineous parents, born weighing 2.3 kg, and a cleft palate repaired at 15 months. There was no history of hypoglycemia or prolonged neonatal jaundice. She had hypotonia, feeding difficulties, and global developmental delay, attaining major motor milestones by the age of three years. She is now studying in 9th standard and has poor academic performance. Anger issues and emotional lability were also noted by the parents. They had consulted pediatricians on and off from a young age, but no specific diagnosis was made, and no definite management was offered.
On examination, her weight was 55 kg (Z-score +0.53), height was 143 cm (Z-score –2.28), and her body mass index was 26.89 kg/m2. On head-to-toe examination, she had a small mouth, prominent philtrum, depressed chin, truncal obesity [Figure 1], and acanthosis nigricans over the neck and axilla. Her hands and feet were small [Figure 2]. Her sexual maturity rating revealed breast stage 3 and pubic hair stage 3 development. Systemic examination was otherwise unremarkable.

- She had a small mouth, prominent philtrum, depressed chin, truncal obesity, and small hands and feet (Black arrows).

- Hands and feet were strikingly small.
Laboratory investigations revealed normal complete blood counts and renal and liver function tests. The free thyroxine was 0.9 ng/dL, thyroid-stimulating hormone 2.78 mIU/mL, luteinizing hormone 5.11 mIU/mL (NR: 0.4-11.7 mIU/mL), follicle stimulating hormone 5.5 mIU/mL (NR: 1-9.2 mIU/mL) and serum estradiol 40 pg/mL (NR: 34-170 pg/mL), 8 am serum cortisol 5.6 µg/dL, fasting blood sugar 90 mg/dL, normal fasting lipid profile, and serum prolactin 20 ng/mL. X-ray left wrist and hand revealed epiphyseal fusion [Figure 3]. Ultrasonography showed an anteverted uterus measuring 3.1 × 2.2 × 2.4 cm, uterine cervix ratio of 1:1, and bilateral ovarian volume of 1.7–2 cc [Figure 4]. The karyotype was 46, XX. A provisional diagnosis of Prader–Willi syndrome (PWS) was considered based on the history of hypotonia at birth, behavioral issues, childhood-onset weight gain, and characteristic facial features. A methylation study targeting deletion/duplication revealed 100% methylation in 7 probes within SNRPN and MAGEL2, confirming a pathogenic variant of PWS due to uniparental disomy.
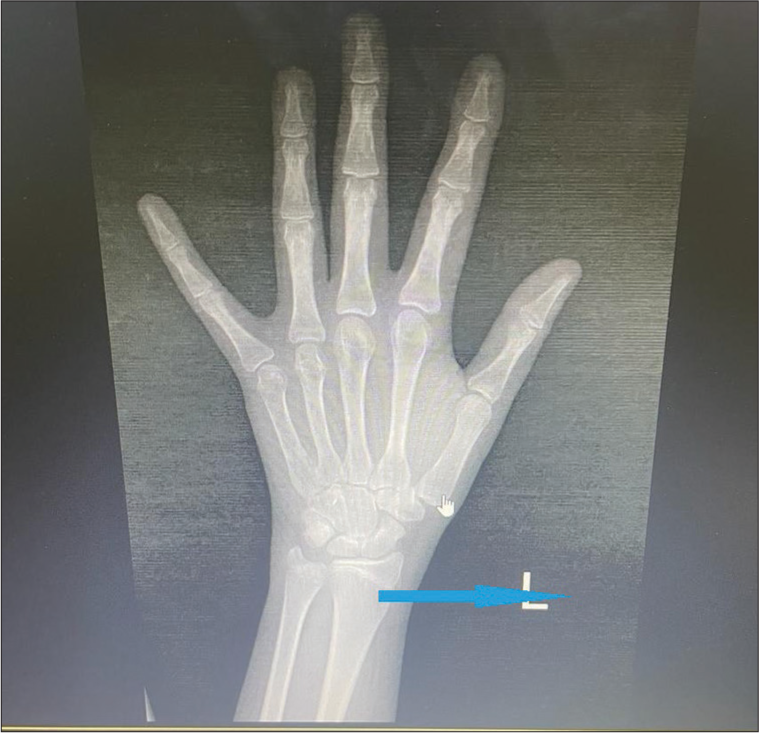
- X-ray left wrist and hand revealed epiphyseal fusion (Blue arrow).
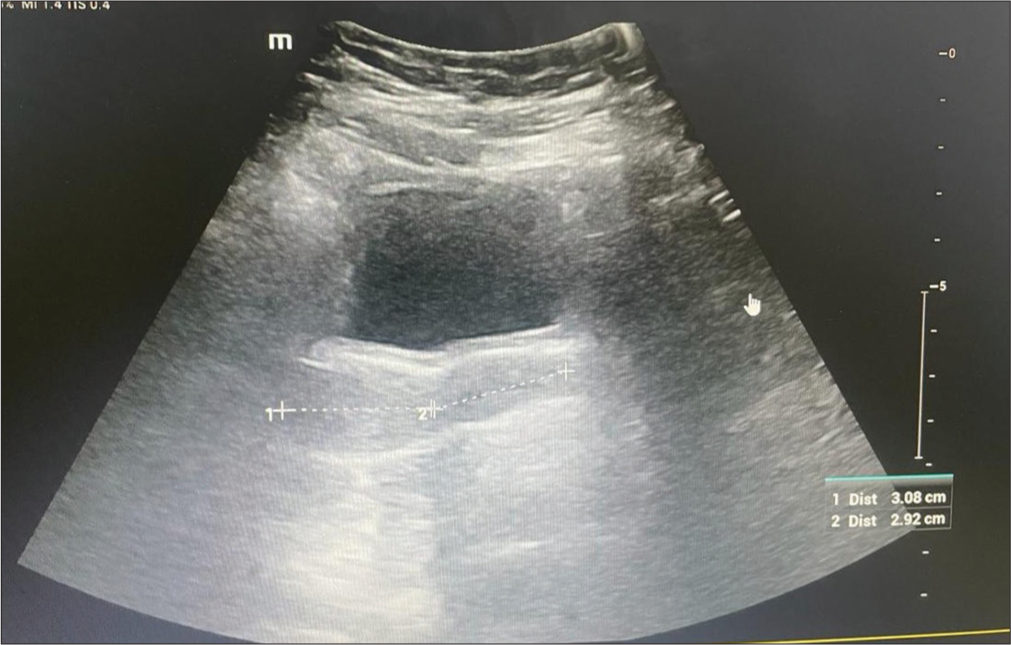
- Ultrasonography showed an anteverted uterus measuring 3.1 × 2.2 × 2.4 cm, uterine cervix ratio of 1:1, and bilateral ovarian volume of 1.7–2 cc.
DISCUSSION
Intellectual incapacity, behavioral issues, hypothalamic dysfunction, and particular dysmorphisms are the hallmarks of PWS. Growth hormone (GH) insufficiency, hypogonadism, and an energy balance dysregulation linked to hypoactivity and insatiable hunger, which leads to a rise in body fat and a decrease in muscle mass, are all consequences of hypothalamic dysfunction. Early initiation of GH therapy not only has a positive action on linear growth but also improves body composition, physical strength, and cognitive level and ameliorates the phenotypic appearance of the syndrome.
Hypogonadism is typically hypogonadotropic in PWS and is generally attributed to hypothalamic-pituitary dysfunction. While hypogonadism in males with PWS is evident at birth, it is difficult to recognize in young PWS females. In most girls, this condition is less noticeable because breast development and maturation are nearly complete. Although primary amenorrhea is frequent, spontaneous menarche can occur late in some cases. Bleeding in these girls could occur as a result of endometrial estrogenization brought on by estrogen produced by peripheral conversion of adrenal androgens. However, one-third of PWS girls who experienced spontaneous menarche had secondary amenorrhea.[1,2]
CONCLUSION
Hypogonadism in PWS is not uncommon. We want to highlight that the diagnosis of PWS was overlooked during infancy and childhood. This oversight could have been prevented with greater awareness among treating clinicians regarding the clinical features and course of the syndrome.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Prader-Willi syndrome and hypogonadism: A review article. Int J Mol Sci. 2021;22:2705.
- [CrossRef] [PubMed] [Google Scholar]
- Hypogonadism in the PraderWilli syndrome from birth to adulthood: A 28-year experience in a single centre. Endocr Connect. 2021;10:1134-46.
- [CrossRef] [PubMed] [Google Scholar]




