
Translate this page into:
Genetics for the pediatric endocrinologists – 2 Primordial short stature in children and adolescents

-
Received: ,
Accepted: ,
How to cite this article: Gupta AK, Gupta N. Primordial short stature in children and adolescents. J Pediatr Endocrinol Diabetes 2022;2:68-77.
Abstract
Primordial short stature (PSS) is an extremely rare group of monogenic disorders characterized by profound global growth failure commencing in the fetal life and continuing postnatally for the rest of the life. It is suspected when there is an extreme degree of proportionate short stature and global growth failure with or without microcephaly, and without any evidence of known skeletal dysplasia. The cardinal features of PSS include severe intrauterine and postnatal growth failure, severe postnatal short stature, primary microcephaly, characteristic facial features, and variable intellectual disability. The most common genetic etiology is monogenic except for Silver–Russell syndrome, where an epigenetic mechanism is a predominant cause of primordial dwarfism. This review demonstrates a holistic approach to the diagnosis and management of PSS in children and adolescents.
Keywords
Cell cycleopathy
Microcephaly
Primordial short stature
Seckel syndrome
Silver–Russell syndrome

CASE SCENARIOS
Case 1
A 5-year-old girl, firstborn of third-degree consanguineous parents, presented with a history of poor growth and limb length asymmetry. She was born at term gestation by normal vaginal delivery at home and cried immediately at birth; her birth weight was 1.4 kg. She was admitted to the neonatal care unit for low birth weight and discharged after 10 days without any requirement for intensive care when regular feeding was established. The third-trimester antenatal ultrasound scan was suggestive of intrauterine growth restriction (IUGR). There was no history of maternal illnesses or exposure to drugs during the antenatal period. Parents noticed persistent feeding difficulty throughout infancy. There was no history of chronic illnesses, seizures, or hearing or vision deficits. Her younger siblings were healthy and growing at age-appropriate percentiles, and family history was not significant. She had a borderline delay in motor milestones. Her height at the time of presentation was at –3.5 SD and her head circumference was –0.9 SD. On general physical examination, the forehead was prominent with a small chin [Figure 1a] and bilateral clinodactyly of the fifth fingers. There was proportionate short stature with limb length asymmetry with the right side limbs larger than the left side. The systemic examination was within normal limits. The skeletal survey was unremarkable. She was later diagnosed as a case of Silver–Russell syndrome (SRS) by methylation studies [Figure 1b]. A multidisciplinary approach to management was instituted including nutritional therapy, early stimulation, physical, and occupational therapy. Growth hormone (GH) therapy with regular growth monitoring was also started.
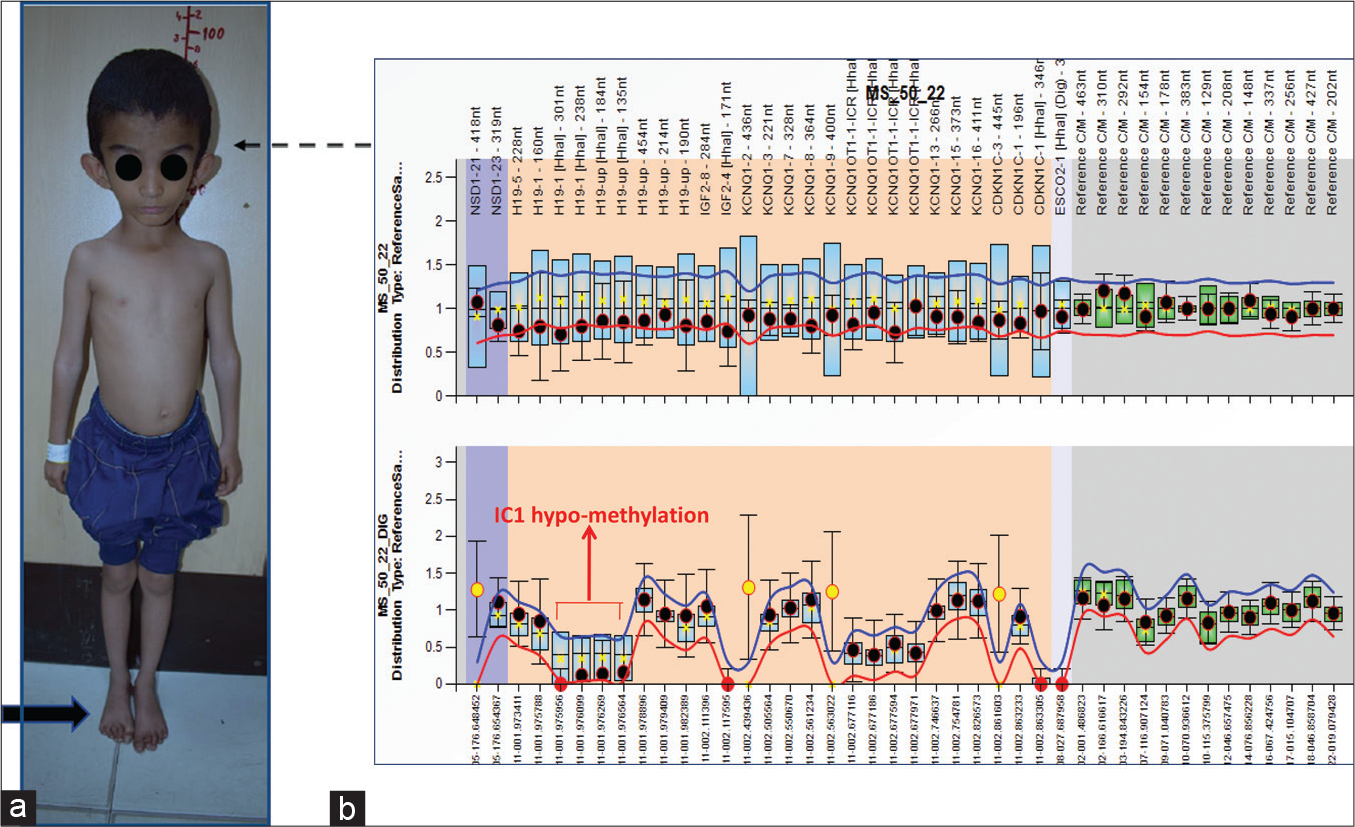
- (a) A 5-year-old girl with Silver–Russell syndrome (SRS, Case 1). Note the short stature, relative macrocephaly, small triangular face (interrupted black arrow), and limb asymmetry (solid black arrow). (b) MS-MLPA methylation ratio result shows IC1 hypomethylation in SRS patient. The specific probes are marked with arrow, IC1: Imprinting centre1.
Case 2
A 6-month-old male infant, a product of non-consanguineous marriage, was brought with poor growth. The growth retardation was detected antenatally when the antenatal fetal anomaly scan was suggestive of IUGR. He was delivered at term by lower segment cesarean section with a birth weight of 1.6 kg with an uneventful perinatal transition. The baby continued to have retarded growth postnatally and at 6 months and weighted had weight at –6.3 SD, head circumference at –5.1 SD, and length at –4.1 SD with proportionate short stature. His developmental milestones at 6 months were age appropriate. On general physical examination, he had a squeaky voice, a prominent nose with broad nasal bridge and hypoplastic alae nasi, micrognathia, and prominent cheeks [Figure 2]. He was clinically diagnosed as a case of microcephalic primordial short stature (PSS). The exome sequencing showed a variant in the pericentrin gene (PCNT) confirming the diagnosis of microcephalic osteodysplastic primordial dwarfism type 2 (MOPD-2). Multidisciplinary symptomatic management was initiated with regular monitoring of growth and development along with surveillance for potential skeletal deformities, diabetes mellitus, hypertension, dental disease, cerebrovascular complications, and learning issues. Genetic counseling was provided and the option for prenatal testing in future pregnancy was provided in view of the recurrence risk of 25% in each pregnancy.
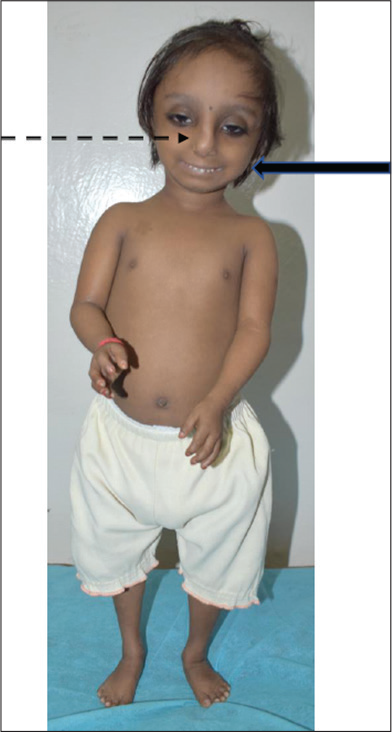
- Case 2. MOPD-2, Note prominent nose, elevated broad nasal root and bridge, hypoplastic ala nasi (interrupted arrow), and micrognathia with prominent cheeks (solid arrow)
Both the above two cases were diagnosed to have PSS and the diagnosis was confirmed by genetic studies. In this review, the authors discuss the definition, etiology, clinical features, approach for genetic testing, and management of PSS.
INTRODUCTION
Growth begins at conception and continues throughout life at a variable pace (the most rapid being the intrauterine phase with variable deceleration postnatally) under the influence of genetic, hormonal, and environmental factors. The genetic control of growth involves the development and function of the pituitary gland, regulation of GH/ insulin-like growth factor (GH/IGF) axis, downstream of GH/IGF1 action, and various other factors involved in the cascades of intracellular processes, including cell cycle regulation. Decreased number of total cells in the body is considered the primary pathogenic mechanism responsible for PSS.
PSS is an extremely rare group of monogenic disorders characterized by profound global growth failure commencing in the fetal life and continuing postnatally for the rest of the life. The cardinal features of PSS include severe intrauterine and postnatal growth failure, severe postnatal short stature, primary microcephaly, characteristic facial features, and variable intellectual disability.
More than 600 genetic variants associated with human growth are identified using genome-wide association studies.[1-3] Monogenic genetic etiology is responsible for the majority of PSS disorders except for SRS, where an epigenetic mechanism is the most common etiology. The causative genes for microcephalic PSS are mainly involved in cellular processes that regulate the cell cycle, replisome, DNA damage, mitosis kinetics and checkpoint control, centrosome cycle, and mitotic spindle functioning.[4,5]
Recently, single-gene variants, especially those involved as components of the DNA replication machinery and DNA repair (e.g., DNA2 and RBBP8/CTIP), and factors required for replication fork stability (e.g., TRAIP and DONSON) have been identified by next-generation sequencing (NGS)-based technology as causative factors in PSS.[6-9] With the advent of the RNA sequencing approach, an aberrant splicing mechanism associated with DONSON has also been discovered.[10] Figure 3 shows a diagrammatic representation of cell cycleopathy genes associated with PSS.
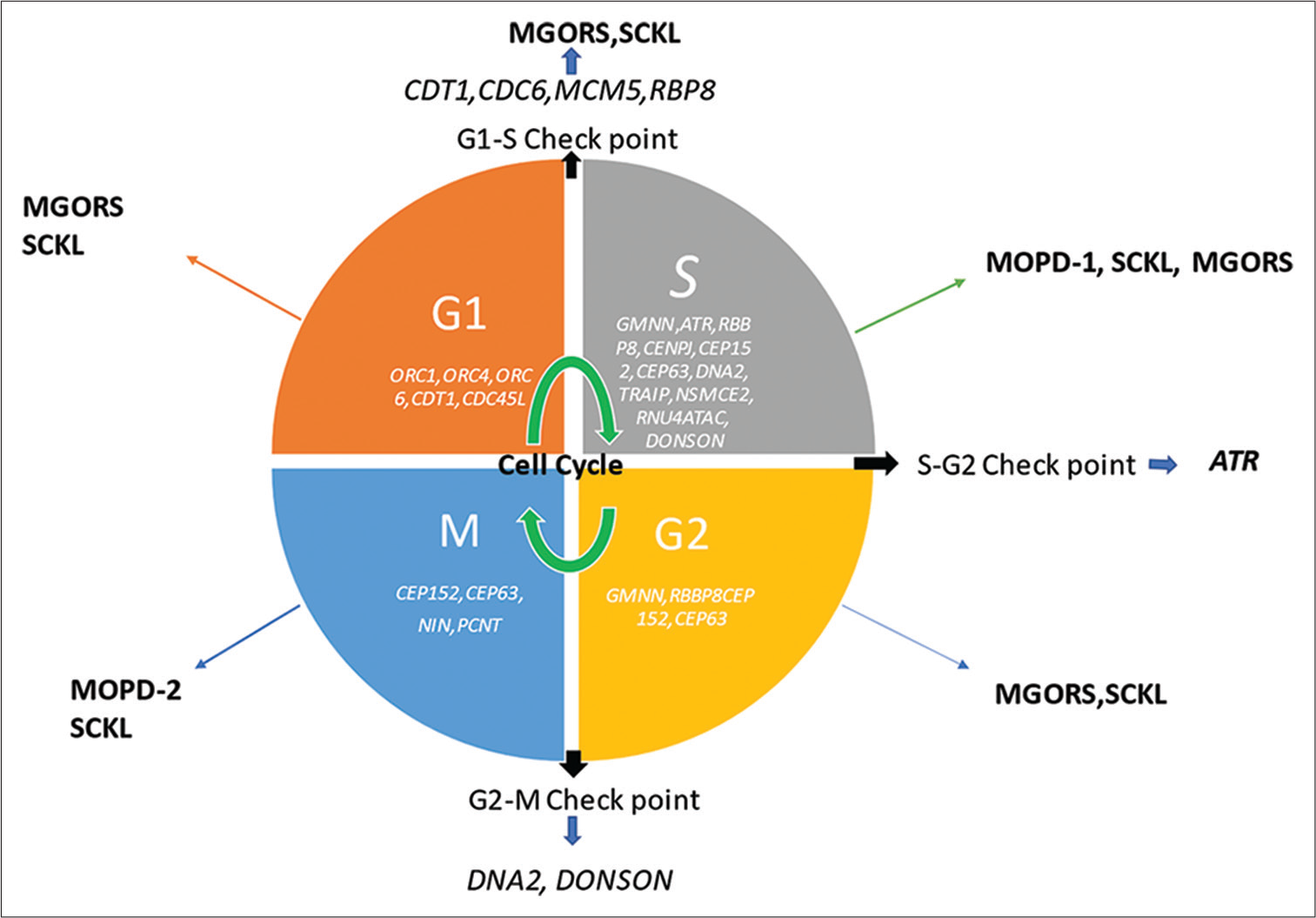
- Cell cycleopathy genes associated with primordial short stature; ATR: Ataxia telangiectasia and Rad3-related protein, G1 phase: First growth phase of cell cycle, G2: Second growth phase of cell cycle, M-Phase: Mitotic Phase of cell cycle, MOPD: Microcephalic osteodysplastic primordial dwarfism, MGORS: Meier–Gorlin syndrome, S phase: Synthesis phase of cell cycle, SCKL: Seckel syndrome, SRS: Silver–Russell syndrome, Gene names are written in italics.
What are common forms of PSS presenting during childhood and adolescence?
Clinically, PSS can be classified into two categories based on the presence or absence of microcephaly as shown below.
PSS with microcephaly – Seckel syndrome (SCKL), MOPD, and Meier-Gorlin syndrome (MGORS)
PSS without microcephaly – SRS and three M (3-M) syndrome.
As mentioned above, based on the genetic mechanisms, PSS can be classified as epigenetic (e.g., SRS) and non-epigenetic in origin (e.g., SCKL, MOPD, MGORS, and 3-M syndromes). In the current review, we have focused on the commonly encountered PSS syndromes.
When do we clinically suspect PSS in a child presenting with poor height gain?
PSS is suspected when there is an extreme degree of proportionate short stature and global growth failure with or without microcephaly, and without any evidence of known skeletal dysplasia. Individuals with PSS have a reduction in head size in proportion to or to a greater extent than the reduction in their body size. This feature differentiates PSS from other forms of short stature.
What are the common syndromes of PSS? What are their cardinal features?
The common syndromes associated with PSS are given and the clinical clues for various types of PSS are listed in [Table 1]. A short description of the common syndromes is given below.
| Disorder (OMIM number) | Gene (s) | Salient clinical features | Inheritance | Reference |
|---|---|---|---|---|
| Imprinting disorder | ||||
| Silver-Russell syndrome (180860) |
• Hypomethylation at 11p15.5 H19/IGF2-imprinting control region (ICR1) (40–50%) • Alterations of chromosome 7 • Maternal uniparental disomy of Chr 7 (mUPD7) in <10% • 7p11.2-11.3 mat dup) 1% |
• IUGR with persistent postnatal growth deficiency • Triangular face • Normal sized cranium • Incurved (clinodactyly) fifth fingers • Asymmetric limb length(may lead to decreased growth on the affected side with hemi-hypertrophy) • Risk of motor and cognitive developmental delay with learning disabilities |
Sporadic heterogeneous (epigenetic changes) |
12 |
| Monogenic | ||||
| Seckel syndrome (201600, 606744, 613676, 61382, 614728, 614850, 615807, 616777, 617253) |
• SCKL1: ATR(3q23) (ataxia-telangiectasia and RAD3-related) defective ATR-dependent DNA damage signaling • SCKL2: RBBP8(18q11) • SCKL4: CENPJ(13q12); centrosomal protein with regulation of microtubule assembly and nucleation • SCKL5: CEP152(15q21) • SCKL6: CEP63(3q22) • SCKL7: NIN(14q22) • SCKL8: DNA2(10q21) • SCKL9: TRAIP(3p21) • SCKL10: NSMCE2(8q24 |
• IUGR and severe proportionate short stature (postnatal persistence) • Microcephaly • Prominent nose • Micrognathia |
AR | 29, 30, 31, 32, 33, 34, 35, 36 |
| Microcephalic osteodysplastic primordial dwarfism type I (210710) | RNU4ATAC located on chromosome 2q14 | • Severe brain malformations • Microcephaly • Short bowed long bones • Dry skin • Sparse hair • Fatal in the first few years of life |
AR | 18 |
| Microcephalic (Majewski) osteodysplastic primordial dwarfism type 2 (210720) | PCNT(21q22.3) | • IUGR • Severe persistent postnatal proportionate short stature • High forehead • Pronounced, progressive microcephaly • Osteodysplasia • Absent or mild intellectual disability |
AR | 17 |
| MGORS (224690) | • MGORS1: ORC1(1p32.3) (shortest stature than other MGORS mutations) • MGORS2: ORC4(2q23.1) • MGORS3: ORC6(16q11) • MGORS4: CDT1(16q24) • MGORS5: CDC6(17q21) • MGORS7: CDC45L(22q11) • MGORS6: GMNN(6p22) |
• Severe intrauterine and postnatal growth retardation • Bilateral microtia • Microcephaly(variable • Aplasia or hypoplasia of the patellae • Intellect usually normal |
AR and AD (MGORS6: GMNN) | 22, 23, 24, 25 |
| Three M (3-M) syndrome (273750, 612921, 614205) |
• CUL7(6p21.1) • OBSL(2q35) • CCDC(19q13.32 ) |
Facial features Normal mental development Long, slender tubular bones Reduced Anteroposterior diameter of vertebral Bodies Delayed bone age |
AR | 19, 20, 21 |
Cardinal features other than growth retardation are italicized; gene names are italicized in the column 2 Gene (s). OMIM: Online Mendelian Inheritance in Man, SCKL: Seckel syndrome, IUGR: Intrauterine growth restriction, MGORS: Meier–Gorlin syndrome, AR: Autosomal recessive, AD: Autosomal dominant. SRS: Silver–Russell syndrome
SRS
SRS is one of the most common forms of PSS. It is characterized by IUGR, postnatal growth failure, prominence of the forehead with relative macrocephaly, and feeding difficulties [Figure 1a, Case 1]. Clinical features that are more commonly associated with SRS rather than small for gestational age (SGA) include low muscle mass, crowded or irregular teeth, micrognathia, downturned corners of the mouth, clinodactyly, limb asymmetry, and excessive sweating.[11] Although the diagnosis of SRS is primarily clinical, there is wide phenotypic variation among affected individuals. International consensus guidelines recommend the use of the Netchine-Harbison clinical scoring system for the diagnosis of SRS [Box 1].[13] A consensus statement with recommendations for clinical diagnosis, investigation, and management of patients with SRS has been recently published.[12,13]
| SGA (birth weight and/or birth length) | ≤−2 SDS for gestational age |
| Postnatal growth failure | Height at 24±1 months ≤−2 SDS or height ≤ −2 SDS below midparental target height |
| Relative macrocephaly at birth | Head circumference at birth ≥1.5 SDS above birth weight and/or length SDS |
| Protruding forehead | Forehead projecting beyond the facial plane on a side view as a toddler (1–3 years) |
| Body asymmetry | LLD of ≥0.5 cm or arm asymmetry or LLD <0.5 cm with at least two other asymmetrical body parts (one non-face) |
| Feeding difficulties and/or low BMI | BMI ≤−2 SDS at 24 months or current use of a feeding tube or cyproheptadine for appetite stimulation |
Clinical diagnosis is considered if a patient scores at least four out of six from the above criteria if all molecular tests are normal and differential diagnoses have been ruled out, patients scoring at least four of the six criteria, including both prominent forehead and relative macrocephaly should be diagnosed as clinical Silver–Russell syndrome BMI: Body mass index, LLD: Leg length discrepancy, SDS: Standard deviation score, SGA: Small for gestational age
SRS is almost always sporadic, and a molecular etiology can be established in up to 60% of cases. Half of these cases are due to hypomethylation of the paternal allele of the H19 gene on chromosome 11p15. Maternal uniparental disomy (complete or segmental) for chromosome 7 accounts for 5–10% of cases; other genetic causes include variable duplications of the critical region on the maternal allele of chromosome 11p15.5, loss of function mutations at IGF2 on the paternal chromosome, and gain of function mutations at CDKN1C. Molecular diagnosis in the majority of cases can be established by DNA methylation specific multiplex ligation-dependent probe amplification (MS-MLPA), as shown in [Figure 1b].
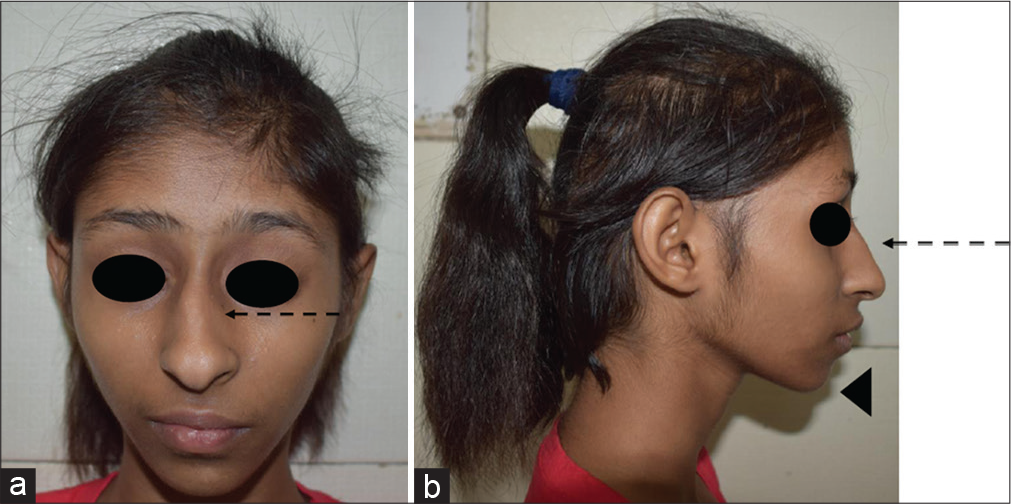
- (a and b) A child with Seckel syndrome. Note the sloping forehead, high nasal bridge with prominent beak-like nose (interrupted arrow), a narrow face, large ears, and micrognathia (arrow head).
GH therapy not only increases final height but also improves body composition and motor development and decreases the risk of hypoglycemia.[14]
SECKEL SYNDROME
This is another common form of PSS. It is genetically heterogeneous and is characterized by dysmorphic features such as a sloping forehead, high nasal bridge with prominent beak-like nose, a narrow face, large ears, and micrognathia [Figure 4].[15] They have moderate-to-severe intellectual disability, hyperactivity, aggressive behavior, and seizures. The phenotypic features are indistinguishable for different genes. Cerebral atrophy and gyral simplification may be seen in neuroimaging.[16] It has phenotypic overlap with MOPD-2, which has more symmetric global growth failure. Several genes [Table 1] are associated with Seckel syndrome; therefore, NGS-based exome sequencing may be used for the identification of the causative variant.
MOPD-2
These babies are extremely small at birth, with progressive development of short stature with relative preservation of head size. Truncal obesity in childhood soon follows the lean and thin body habitus of infancy. Feeding issues are more common in infancy, while refractory errors in vision and sensorineural hearing loss develop over time. Dyslipidemia, cardiomyopathy, and type 2 diabetes mellitus develop at a young age. Craniofacial features include craniosynostosis, prominent nose, elevated broad nasal root and bridge, hypoplastic alae nasi, micrognathia, prominent cheeks, low set simple ears with lack of lobule, relatively large eyes with downslanting palpebral fissures, shallow orbits, enamel hypoplasia, and oligodontia [Figure 2, Case 2]. Skeletal findings include scoliosis and progressive ligamentous laxity resulting in radial and patellar dislocation over time. Imaging findings include long gracile bones, delayed epiphyseal ossification, severe coxa vara, metaphyseal flaring, high narrow ilia, flat acetabular angles, mild platyspondyly, 11 ribs, pseudoepiphyses of metacarpals, small facial bones, large sella, abnormal brain myelination, ventriculomegaly, and hypoplasia/cyst of the corpus callosum. It can be differentiated from Seckel syndrome by more severe retardation of growth and less pronounced microcephaly and intellectual disability. Pericentrin, PCNT, is the gene involved with an autosomal recessive (AR) inheritance pattern.[17]
MOPD-1
This syndrome overlaps clinically with other types of primordial dwarfism; however, it is differentiated by the presence of brain malformations and early lethality with death before 28 months of age. Patients with MOPD-1 have brachycephalic body proportions and severe microcephaly. Craniofacial findings include a sloping forehead, prominent beaked nose, protruding eyes, micrognathia, and sparse eyebrows. Other abnormalities include platyspondyly, cleft vertebral arches, delayed epiphyseal ossification, low broad dysplastic pelvis, poor acetabular formation, short broad bowed humeri and femora, unremarkable metaphyses, agenesis of the corpus callosum, colpocephaly, marked lissencephaly, heterotopia and other neuromigrational defects, vermis agenesis, and arachnoid cyst. MOPD-1 is caused by genetic variants in the RNU4ATAC gene, which codes for small nuclear RNA component of the U12 spliceosome, required for the proper excision of the U12-dependent class of introns.[18]
OTHER DNA REPAIR DEFECTS
Bloom syndrome is characterized by growth retardation, photosensitivity, erythematous “butterfly rash,” and a high rate of bacterial infections due to immunodeficiency.
Cockayne syndrome is characterized by low to normal birth weight, postnatal growth failure, prematurely aged appearance with deep-set eyes and a pinched appearance to the nose (loss of subcutaneous fat), cutaneous photosensitivity, pigmentary retinopathy and/ or cataracts and/or optic atrophy, and sensorineural hearing loss, and brain dysmyelination with calcium deposits.
Nijmegen breakage syndrome is characterized by microcephaly, growth retardation, prominent midface, and mild learning difficulties.
Rothmund–Thomson syndrome is phenotypically distinct and suspected in the presence of specific features such as growth deficiency, poikiloderma congenita, photosensitivity, radial ray defects in 25%, dystrophic nails, abnormal teeth, and hypogonadism.
THREE M (3-M) SYNDROME
3-M syndrome is another PSS that is commonly referred to for genetic evaluation. It is characterized by prenatal and postnatal growth retardation, short neck, and long slender bones. Dysmorphic features include dolichocephaly, frontal bossing, triangular face, malar hypoplasia, full and pointed chin, fleshy nasal tip, short nose with anteverted nares, long philtrum, full lips [Figure 5a], prominent heels [Figure 5b], and delayed dentition with normal intelligence.[19] Variable skeletal findings on the skeletal survey include long slender bones [Figure 5c], winged scapulae, tall vertebrae, and joint hypermobility. Serum levels of GH and IGF1 are normal. Genes involving ubiquitin-proteasome pathway, for example, CUL7 (65%), OBSL1 (30%), and CCDC8 (5%) are implicated in AR manner.[20,21]
MGORS
MGORS is characterized by a triad of microtia, patellar aplasia/hypoplasia, and short stature.[22] Additional frequent clinical features are mammary hypoplasia and abnormal genitalia (predominantly cryptorchidism and hypoplastic labia minora/majora). The majority have normal intellect with proportionate microcephaly (95% of the cases have normal IQ) and a cheerful, friendly personality.[23] Growth failure is variable and can be mild. Mammary hypoplasia is found in all post-pubertal females, with nipple hypoplasia and sparse axillary and pubic hair. Infantile phenotype with micrognathia, small mouth and full lips gradually changes over time into a phenotype with a high vertical forehead, narrow nose, and high nasal bridge. Treatment with GH and estrogen is associated with improvement in growth retardation and hypoplasia of the breast and genitals. Severe congenital pulmonary emphysema can be life-threatening. It follows both AR and autosomal dominant (AD) inheritance patterns. AR mutations in ORC1, ORC4, ORC6, CTD1, CTC6, MCM5, and CDC45 genes encode for components of the pre-replication complex during DNA replication and AD mutations in GMNN encode protein inhibiting DNA replication by binding to DNA replication factor, Cdt1. Congenital pulmonary emphysema is strongly associated with CDT1 variant.[24-26]
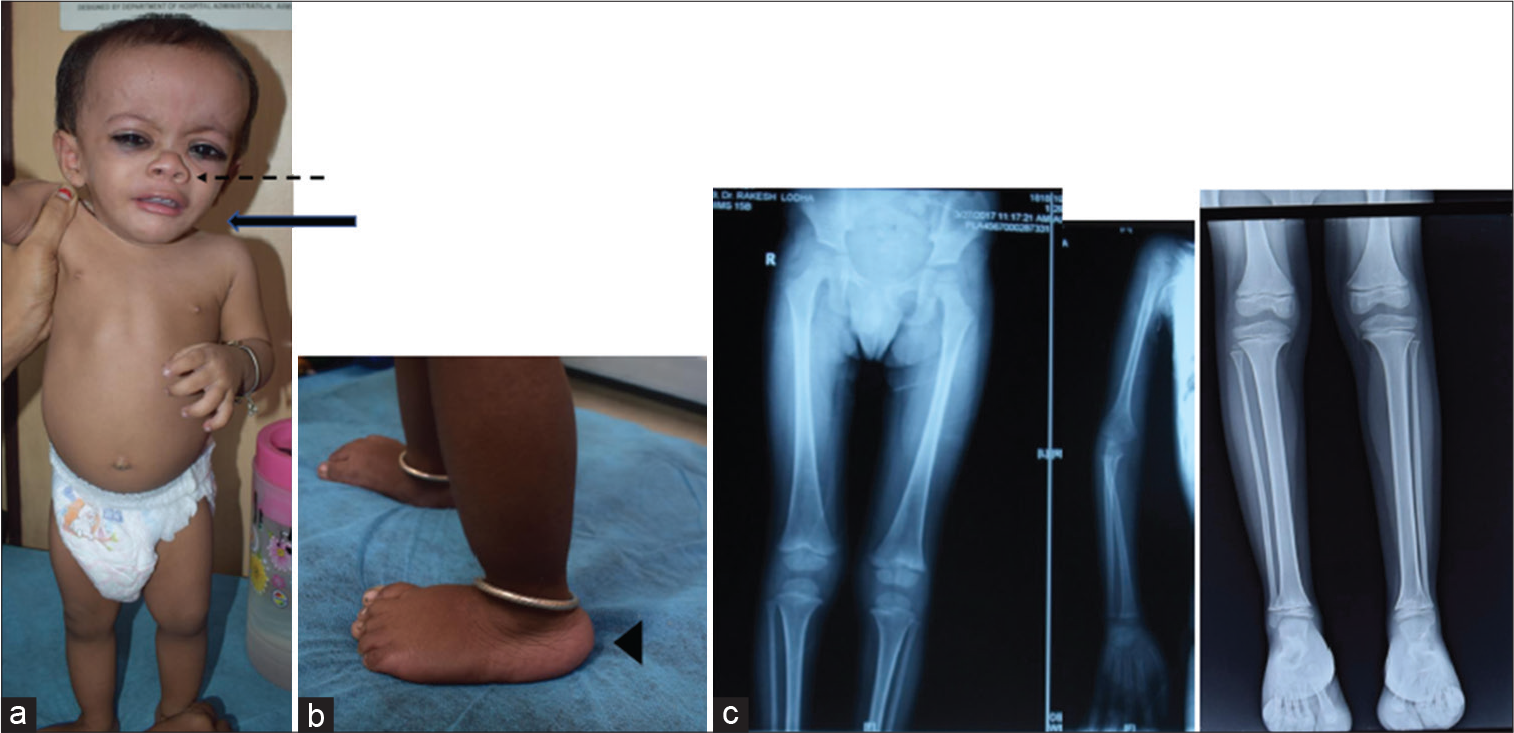
- A child with 3-M syndrome – (a) fleshy nasal tip, upturned alae nasi (interrupted arrow), short neck (solid bold arrow), triangular face with frontal bossing, and full eyebrows, (b) prominent heels (arrow head), and (c) slender shaft of long bones, small pelvis and iliac wings, and short femoral necks.
OTHER PRENATAL ONSET SHORT STATURE SYNDROMES
Dubowitz syndrome
Dubowitz syndrome is an AR condition with pre-and postnatal growth retardation with eczema, telecanthus, blepharophimosis, and broad nasal bridge.
Leprechaunism (Donohue syndrome)
Leprechaunism (Donohue syndrome) is an AR disorder of pre-and postnatal growth failure that is caused by biallelic variants in the insulin receptor gene and carries a poor prognosis. These children have thick lips, hirsutism, clitoromegaly, and acanthosis nigricans with elevated serum insulin (insulin resistance).
IMAGe syndrome
IMAGe is an acronym for IUGR, metaphyseal dysplasia, adrenal hypoplasia, and genitourinary abnormalities (seen only in males). Their facial phenotype mimics SRS with a triangular face, a frontal bossing, depressed wide nasal bridge with micrognathia, or retrognathia. Growth is severely retarded, beginning in the fetal period with preserved head circumference. Uniformly, all cases develop some degree of skeletal abnormality, predominantly affecting metaphysis. Adrenal insufficiency is another universal phenomenon affecting this syndrome. The majority have normal development and cognition.
There are no established criteria for clinical diagnosis of IMAGe syndrome and diagnosis is suspected on clinical grounds and confirmed by the molecular finding of a heterozygous CDKN1C pathogenic variant in the maternal PCNA allele. CDKN1C (expressed in maternal alleles only) is located at domain 2 of 11p15.5 imprinting cluster and regulates cell growth by cell cycle inhibition; gain of function pathogenic variants in CDKN1C leads to over-inhibition of cell cycle resulting in severe growth retardation beginning in fetal life.[27]
It is differentiated from SRS by the absence of clinodactyly of the fifth finger, café-au-lait macules, and limb length discrepancy. Evaluation involves workup for adrenal insufficiency, imaging for skeletal and/or hip dysplasia and genitourinary anomalies in males, and neurological and developmental assessment. Management consists of treatment for adrenal insufficiency, orthopedic intervention if required, occupational, speech, and physical therapy. GH therapy may be recommended for growth failure, even though there are limited studies evaluating its role in IMAGe syndrome.
Mulibrey nanism
It is another rare AR growth condition that is clinically characterized by antenatal and postnatal growth failure, short stature, and facial dysmorphism, for example, dolichocephaly, high broad forehead, low depressed nasal bridge, and small pointed chin, long slender bones with over-tabulation and J-shaped sella turcica, and a strange high-pitched voice. Hepatomegaly, benign yellowish pigmentary changes in the ocular fundi, cutaneous nevi flammae, fibrous dysplasia of long bones, congestive heart failure caused by pericardial constriction, myocardial hypertrophy and myocardial fibrosis, as well as insulin resistance and type 2 diabetes are additional features seen in some of the cases. This disorder has been linked to a higher risk of tumor development. It was initially identified in Finland, which suggests the founder effect. Molecular diagnosis is made by identifying a variant in tripartite motif protein 37, which codes for a RING finger ubiquitin E3 ligase.[28]
What are the different tests/technologies available for genetic diagnosis of children with suspected PSS? How do we choose a specific genetic test/technique for a given child with a suspected genetic form of PSS?
Children with proportionate dwarfism with or without microcephaly should be evaluated for the exclusion of underlying genetic etiology. Accurate phenotyping is one of the most critical steps for deciding diagnostic workup and its interpretation. An appropriate choice of the test is required to end the diagnostic odyssey.
There are two approaches for molecular genetic testing: Targeted testing (single-gene testing by Sanger sequencing or multigene targeted panel using NS) when a particular phenotype is clinically suspected or identified; the second approach is NGS-based exome sequencing/genome sequencing when the phenotype is not discernible [Figure 6]. Cases with characteristic findings are more likely to be diagnosed using gene-targeted testing with the advantage of limiting the identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying condition. However, multigene testing is available from most commercial laboratories through the virtual gene panel. The detection rate using sequencing varies. Deletion-duplication analysis using MLPA or chromosomal microarray may be indicated for some conditions. Whole-genome testing may be helpful when no pathogenic variants in known genes or candidate genes/regions could be identified. However, the amount of data generated is enormous and analysis may be complex. For SRS, MS-MLPA is the test of choice.
How do we counsel the family when a child is diagnosed with a genetic or monogenic form of PSS?
PSS is inherited in an AR manner and if both parents are known to be heterozygous for a pathogenic variant, each sibling of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and pre-implantation genetic testing are possible.
SPECIAL SITUATIONS
SRS
Accurate genetic counseling depends on the underlying molecular cause. 11p15 hypomethylation is associated with low recurrence risk. The risk of recurrence might be as high as 50% in maternally inherited CDKN1C gain of function mutations and paternally inherited IGF2 loss of function. Maternal UPD is associated with a low recurrence and offspring risk.[7] There is a paucity of data regarding the recurrence risk and offspring risk for children with clinically diagnosed SRS though it is considered low.
What are the additional evaluations required for the genetic forms of PSS?
Patients with PSS should receive multidisciplinary care in a center of expertise composed of pediatric endocrinologist, pediatric gastroenterologist, nutritionist, clinical geneticist, craniofacial team, orthopedic surgeon, neurologist, speech and language therapist, and psychologist. Once a diagnosis is established, initial evaluation should include a thorough clinical examination, blood pressure monitoring, dental evaluation, renal ultrasound, echocardiography, neuroimaging, and assessment for signs of insulin resistance, diabetes mellitus, and liver disease. They may develop multiple cerebrovascular disorders over time; therefore, identification of initial disease should not exclude them from future screening. Individuals born with a low birth weight are at increased risk of adult health problems, including coronary heart disease, hypertension, dyslipidemia, insulin resistance, and obesity (metabolic syndrome). Neurocognitive support, early stimulation, and supportive care with physical rehabilitation are very important for holistic management.
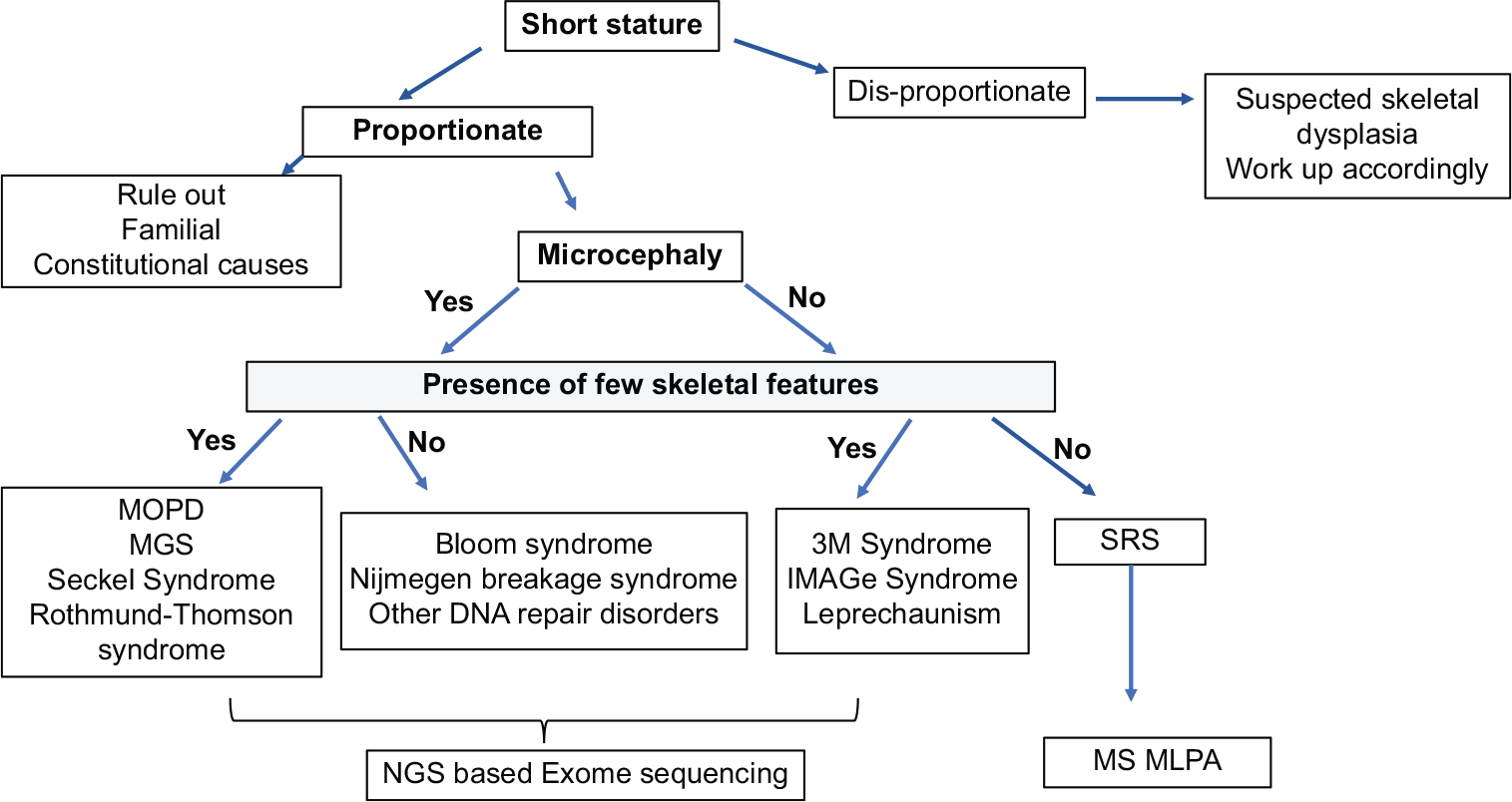
- The genetic testing strategy for PSS. PSS: Primordial short stature, MOPD: Microcephalic osteodysplastic primordial dwarfism, MGORS: Meier-Gorlin syndrome, SRS: Silver–Russell syndrome, MLPA: Multiplex ligation-dependent probe amplification, NGS: Next-generation sequencing.
What is the role of GH therapy in PSS?
Response to recombinant human GH therapy is typically poor in cases of PSS with the exception of SRS. GH therapy seems to have favorable metabolic effects in children born SGA, including increased lean body mass, reduced fat mass, decreased blood pressure, and an improved lipid profile; but there is risk of losing these effects after discontinuation of therapy. GH therapy is recommended for SRS by the US FDA and the European Medicines Agency. The Dutch longitudinal study demonstrated comparable response to GH in patients with SRS and non-SRS SGA children (mean total height gains of 1.30 standard deviation score [SDS] and 1.26 SDS, respectively); however, the final adult height attained in patients with SRS was lower (mean adult height −2.17 SDS vs. −1.65 SDS for non-SRS children born SGA).[37]
CONCLUSION
Conventionally, growth and height disorders are considered multifactorial in origin; however, a proportion of children with some additional features are suspected to have a monogenic etiology. A thorough clinical evaluation and appropriate genetic testing can help establish the diagnosis and guide in further management.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Hundreds of variants clustered in genomic and biological pathways affect human height. Nature. 2010;467:832-8.
- [CrossRef] [PubMed] [Google Scholar]
- Heritability of adult body height: A comparative study of twin cohorts in eight countries. Twin Res. 2003;6:399-408.
- [CrossRef] [PubMed] [Google Scholar]
- Rare and low-frequency coding variants alter human adult height. Nature. 2017;542:186-90.
- [CrossRef] [PubMed] [Google Scholar]
- Human DNA damage response and repair deficiency syndromes: Linking genomic instability and cell cycle checkpoint proficiency. DNA Repair (Amst). 2009;8:1139-52.
- [CrossRef] [PubMed] [Google Scholar]
- Mitotic spindle multipolarity without centrosome amplification. Nat Cell Biol. 2014;16:386-94.
- [CrossRef] [PubMed] [Google Scholar]
- The role of mitotic kinases in coupling the centrosome cycle with the assembly of the mitotic spindle. J Cell Sci. 2014;127:4111-22.
- [CrossRef] [PubMed] [Google Scholar]
- One to only two: A short history of the centrosome and its duplication. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130455.
- [CrossRef] [PubMed] [Google Scholar]
- The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154-60.
- [CrossRef] [PubMed] [Google Scholar]
- Deconstructing the centriole: Structure and number control. Curr Opin Cell Biol. 2012;24:4-13.
- [CrossRef] [PubMed] [Google Scholar]
- Biallelic variants in DNA2 cause microcephalic primordial dwarfism. Hum Mutat. 2019;40:1063-70.
- [CrossRef] [PubMed] [Google Scholar]
- Hand skeletal maturity and its correlation with mandibular dental development. J Clin Exp Dent. 2014;6:e275-9.
- [CrossRef] [PubMed] [Google Scholar]
- A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015;52:446-53.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and management of Silver-Russell syndrome: First international consensus statement. Nat Rev Endocrinol. 2017;13:105-24.
- [CrossRef] [PubMed] [Google Scholar]
- Metabolic health and long-term safety of growth hormone treatment in Silver Russell syndrome. J Clin Endocrinol Metab. 2017;102:983-91.
- [CrossRef] [PubMed] [Google Scholar]
- "Bird headed dwarfs: Studies in Developmental Anthropology Including Human Proportions" Springfield, Ill: CC Thomas Publishers; 1960.
- [Google Scholar]
- CtIP mutations cause Seckel and Jawad syndromes. PLoS Genet. 2011;7:e1002310.
- [CrossRef] [PubMed] [Google Scholar]
- Striking hematological abnormalities in patients with microcephalic osteodysplastic primordial dwarfism Type II (MOPD II): A potential role of pericentrin in hematopoiesis. Pediatr Blood Cancer. 2014;61:302-5.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science. 2011;332:238-40.
- [CrossRef] [PubMed] [Google Scholar]
- The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. Am J Hum Genet. 2009;84:801-6.
- [CrossRef] [PubMed] [Google Scholar]
- Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet. 2011;89:148-53.
- [CrossRef] [PubMed] [Google Scholar]
- Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin Endocrinol (Oxf). 2012;77:335-42.
- [CrossRef] [PubMed] [Google Scholar]
- Meier-Gorlin syndrome genotype-phenotype studies: 35 individuals with pre-replication complex gene mutations and 10 without molecular diagnosis. Eur J Hum Genet. 2012;20:598-606.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in origin recognition complex gene ORC4 cause Meier-Gorlin syndrome. Nat Genet. 2011;43:360-4.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the pre-replication complex cause Meier-Gorlin syndrome. Nat Genet. 2011;43:356-9.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in ORC1, encoding the largest subunit of the origin recognition complex, cause microcephalic primordial dwarfism resembling Meier-Gorlin syndrome. Nat Genet. 2011;43:350-5.
- [CrossRef] [PubMed] [Google Scholar]
- De novo GMNN mutations cause autosomal-dominant primordial dwarfism associated with Meier-Gorlin syndrome. Am J Hum Genet. 2015;97:904-13.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. 2012;44:788-92.
- [CrossRef] [PubMed] [Google Scholar]
- Gene encoding a new RING-B-box-coiled-coil protein is mutated in mulibrey nanism. Nat Genet. 2000;25:298-301.
- [CrossRef] [PubMed] [Google Scholar]
- A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497-501.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel syndrome. PLoS Genet. 2012;8:e1002945.
- [CrossRef] [PubMed] [Google Scholar]
- Novel CENPJ mutation causes Seckel syndrome. J Med Genet. 2010;47:411-4.
- [CrossRef] [PubMed] [Google Scholar]
- CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet. 2011;43:23-6.
- [CrossRef] [PubMed] [Google Scholar]
- Mutation in PLK4, encoding a master regulator of centriole formation, defines a novel locus for primordial dwarfism. J Med Genet. 2014;51:814-6.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet. 2007;40:232-6.
- [CrossRef] [PubMed] [Google Scholar]
- Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science. 2011;332:240-3.
- [CrossRef] [PubMed] [Google Scholar]
- Studies of microcephalic primordial dwarfism I: Approach to a delineation of the Seckel syndrome. Am J Med Genet. 1982;12:7-21.
- [CrossRef] [PubMed] [Google Scholar]
- Long-term results of GH treatment in Silver-Russell syndrome (SRS): Do they benefit the same as non-SRS short-SGA? J Clin Endocrinol Metab. 2016;101:2105-12.
- [CrossRef] [PubMed] [Google Scholar]




